

TEMA I
CÉLULAS OSEAS
Existen tres tipos principales de células óseas: osteoblastos, osteoclastos y osteocitos.
Osteoblastos: Células muy diferenciadas que son las responsables del depósito de la matriz extracelular y su mineralización. Presentan una estructura celular que incluye un gran retículo endoplásmico, complejo de Golgi y características celulares relacionadas con su papel de síntesis de proteínas y de células secretoras. Participan activamente en la formación de hueso.
Osteoclastos: Responsables de la resorción de hueso calcificado y de cartílago, están formados por la fusión de precursores mononucleares. Las células muestran polaridad, ocurriendo la resorción a lo largo del borde rugoso que está situado a nivel de la superficie ósea.
Osteocitos: Se trata de osteoblastos que permanecen por detrás en lagunas a medida que avanza la superficie formadora de hueso. Estas células se comunican entre sí a través de procesos citoplasmáticos que atraviesan los canalículos óseos, que pueden ser de ayuda para coordinar la respuesta del hueso a las fuerzas o a la deformación.
TEMA II
Células de los huesos
Al considerar las células de los huesos es necesario diferenciar los elementos que pertenecen estrictamente al hueso de aquellos que pertenecen a la médula ósea. Aunque los progenitores de los osteoclastos son células hematopoyéticas también son consideradas como célulás óseas. Por consiguiente, se consideran como células óseas las células progenitoras, los osteoblastos, los osteocitos, las células tapizantes del hueso (denominadas por los anglosajones "bone lining cells" y los osteoclastos

Células osteoprogenitoras
Son unas células no especializadas, derivadas del mesénquima que pueden experimentar mitosis y transformarse en osteoblastos . Estas células se encuentran en la parte interna del periostio, en el endostio y en los canales perforantes y de Havers. Ocasionalmente y bajo la influencia de factores de crecimiento como el TGFb (factor de crecimiento transformante b) algunas células hematopoyéticas de la médula ósea pueden diferenciarse a células osteoprogenitoras

Los osteoblastos son las células responsables de la formación y organización de la matriz extracelular del hueso y de su posterior mineralización. Además liberan algunos factores que son probablemente mediadores de la resorción ósea.
Son células cuboides que forman una capa en las superficies de los huesos en crecimiento, o como en el caso de la osificación intramembranosa, rodean áreas de osificación. Parte de su membrana se encuentra en contacto con el borde osteide, llamándose así el área donde está teniendo lugar la calcificación . Como otras células que fabrican activamente proteínas, los osteoblastos tienen abundante retículo endoplásmico rugoso y un área de Golgi muy desarrollada. Se reconocen fácilmente vesículas de pinocitosis cerca de la membrana responsables de la secreción del colágeno.
El principal producto de los osteoblastos maduros es el colágeno de tipo I que constituye el 90% de las proteínas del hueso. Pero, además, producen otras proteínas como la osteocalcina y las proteínas Gla matriciales, y glicoproteínas fosforiladas incluyendo las sialoproteínas I y II, la osteopontina y la osteonectina. Las principales proteínas con actividad enzimática producidas por los osteoblastos son la fosfatasa alcalina y la colagenasa.

Osteocitos
Una cierto número de osteoblastos quedan atrapados en las lagunas de la matriz, pasando a ser osteocitos. Los osteocitos están interconectados por un sistema de canalículos aunque ya no excretan materiales de la matriz. Los osteocitos pasan por varias fases de maduración hasta que quedan completamente rodeados por la matriz y se mantienen en un estado de aparente reposo. La fase formativa es la que tiene lugar cuando todavía mantienen una actividad osteoblástica quedando atrapados en un tejido parcialmente osteoide. La fase de resorción corresponde a un período de la vida del osteocito en la que es capaz de resorber la matriz ósea del borde de su laguna (fase osteolítica) y, finalmente, en la fase degenerativa caracterizada por picnosis y fragmentación del núcleo los ostocitos probablemente muere. Se desconocen las causas de la degeneración de los ostecitos.

Células tapizantes del hueso
Las superficies inactivas del hueso están cubiertos por una capa de células planas muy delgadas similares a las células endoteliales. Al parecer derivan de los osteoblastos (mantienen una actividad de fosfatasa alcalina) pero se desconoce cuales son sus funciones. Se cree que su papel más importante es separar el fluído intersticial de los fluídos del hueso y contribuir a mantener las concentraciones de calcio

Las células responsables de resorción de la matriz ósea son los osteclastos, células polinucleadas de gran tamaño que se localizan en las superficies óseas firmemente asociadas a la matriz óseo. Los osteoclastos se forman por la fusión de varias células mononucleares derivadas de una célula madre sanguínea de la médula ósea mostrando muchas propiedades de los macrófagos
Los osteoclastos se caracterizan por disponer de una porción de su membrana "arrugada" ,en forma de cepillo, rodeada de un citoplasma libre de orgánulos, llamada "zona clara" con la que se adhiere a la superficie del hueso mediante integrinas, unos receptores especializados del hueso. El proceso de resorción se inicia cuando el aparato de Golgi de la células excreta lisosomas con enzimas capaces de producir un microambiente ácido por debajo de la membrana arrugada como consecuencia del transporte de protones mediante la bomba de protones ATP-dependiente, el intercambio Na+/H+ y la anhidrasa carbónica. Las enzimas lisosomales de los osteoclastos implicadas en este proceso son cistein-proteasas como la catepsina y sobre todo, la fosfatasa ácida tartrato-resistente (esta última se utiliza como marcador del fenotipo osteoclástico). Las enzimas lisosomales solo son liberadas en la zona clara en las proximidades del borde arrugado produciendose en este área las reacciones de degradación de la matriz que deben producirse antes de que le medio ácido disuelva las sales minerales del hueso.
La resorción osteoclástica depende de una serie de factores reguladores externos como la hormona paratiroidea, la 1,25-dihidroxivitamina D3 y la calcitonina. Otros factores que afectan la funcionalidad de los osteoclastos son los glucocorticoides y las prostaglandinas.
omposición de la matriz ósea
El hueso es un tejido heterogéneo, altamente anisótropo de tal manera que la matriz intercelular muestra una estructura y propiedades diferentes en los diferentes tipos de hueso
Constituyentes orgánicos:
El principal componente de la matriz ósea es el colágeno tipo I que supone entre el 90 y 95% de la matriz orgánica. Las fibrillas de colágeno son similares a las que se presentan en otros tejidos y están distribuídas aleatoriamente formando un entramado.
El siguiente producto en importancia es la osteonectina, una fosfoproteína que puede interaccionar tanto con el colágeno como con las sales inorgánicas. Es una proteína altamente reactiva que se localiza preferentemente en las áreas de mayor grado de calcificación. La osteonectina (también llamada SPRC o BM-40 (Secreted Protein Cystein Rich) está codifica por el gen 5q31.3-q32. Su expresión descontrolada está asociada a diversos tipos de cáncer. La osteocalcina (también llamada Proteína Gla) se caracteriza por la presencia de tres residuos de ácido g-carboxiglutámico.
Otras proteínas no colagenosas son la osteopontina (también llamada sialoproteína I) que se une a la hidroxiapatita y es producida por los ostoblastos estimulados por la 1-a-1,25-dihidroxivitamina D, las proteínas óseas morfogenéticas (BMPs) que juegan un papel similar al de los factores de crecimiento y los proteoglicanos ácidos que se encuentran en concentraciones mayores en el área osteide en comparación con la matriz calcificada
Constituyentes inorgánicos:
la matriz ósea contiene abundantes sales minerales en forma cristalizada, en particular la hidroxiapatita o fosfato tricálcico [Ca3(PO4)2.(OH)2] y algo de carbonato cálcico. En cantidades pequeñas se encuentran los sulfatos, fluoruros e hidróxido de magnesio. Todas estas sales se encuentran depositadas en una retícula formada por las fibras de colágeno. El proceso por el cual estas sales se depositan y cristalizan en la retícula se denomina calcificación. Aunque la dureza del hueso se debe a sus componentes minerales, sin la existencia de la retícula de colágeno, el hueso sería frágil. Las fibras de colágeno y otras proteínas presentes en la matriz aportan flexibilidad, resistencia a la tensión. Si faltan las fibras de colágeno o están son defectuosas se producen enfermedades como la osteogénesis imperfecta, también conocida como huesos de cristal. Por contrario, si se eliminan las sales minerales del hueso por disolución en un ácido débil como el vinagre, resulta una estructura flexible, esponjosa y gomosa.
El hueso no es completamente macizo, sino que muestra muchos espacios entre sus componentes duros. Por estos espacios discurren los vasos sanguíneos que nutren las células óseas y reducen el peso del hueso. Según el tamaño y naturaleza de estos espacios, los huesos se denominan compactos o esponjosos.

REGULACION DE LA BIOLOGIA OSEA
El hueso es un tejido dinámico que necesita estímulos para su desarrollo normal y cambia sus características biomecánicas por remodelación.
El tejido óseo sufre un recambio constante denominado remodelación ósea, que permite que el hueso responda a mediano y largo plazo a las necesidades mecánicas y metabólicas del organismo. La integridad del esqueleto requiere que los procesos de formación y reabsorción ósea sean realizados de forma coordinada por las células óseas.
La centellografía ósea refleja los cambios en la actividad metabólica ósea en los que esta basada su alta sensibilidad y precocidad diagnóstica.
La secuencia de procesos fisiopatológicos inducidos o asociados a distintos factores etiológicos involucra varios mecanismos de regulación, entre los que se destacan elementos mecánicos, locales y hormonales.
El remodelado óseo se lleva a cabo mediante los procesos de formación (realizados por el osteoblasto) y de reabsorción ósea (realizados por el osteoclasto).
Los osteoblastos se hallan en contacto directo con las superficies óseas formando un grupo firme de una sola capa de espesor, sintetizan el componente orgánico de la matriz ósea y controlan el depósito de las sales minerales; pasan sucesivamente por tres estadios funcionales, el primero de proliferación celular y síntesis de los componente orgánicos de la matriz ósea (colágeno tipo I, proteoglicanos, osteocalcina y factores de crecimiento), el segundo maduración de la matriz ósea y el ultimo de depósito de mineral. Estas células se hallan en contacto entre sí y con las de las células de la superficie mediante finos canales ínter citoplasmáticos que recorren la matriz ósea y posibilitan una comunicación química y eléctrica, permitiendo el paso directo de una a otra célula de iones inorgánicos, aminoácidos, azúcares, nucleótidos, vitaminas y pequeñas moléculas hidrosolubles. Los osteocitos mantienen las propiedades biomecánicas del tejido óseo, detectando alteraciones mecánicas y microlesiones de la matriz.
La hormona paratiroidea (PTH) es el factor sistémico que más influye en la diferenciación y proliferación de las células del linaje osteoblástico y por un mecanismo autocrino lo hacen numerosos factores locales entre los que se destacan la interleucina-1, el factor de necrosis tumoral (TNF) y los factores de crecimiento.

Los osteoclastos son células multinucleadas, de gran tamaño (derivan de la fusión de los pre-osteoclastos), y poseen anhidrasa carbónica y fosfatasa ácida; se disponen sobre las superficies óseas de manera aislada o en pequeños grupos. El área de adhesión de los osteoclastos a la matriz ósea crea un microambiente ácido que permite solubilizar el componente mineral y digerir la matriz orgánica (enzimas proteolíticas y colagenasas).
Los osteoclastos poseen receptores específicos para la calcitonina pero carecen de receptores para la hormona paratiroidea (PTH). A nivel local la osteoclastogénesis parece ser activada por la IL-6 e IL-11 (segregadas por los osteoblastos) cuya producción es estimulada por hormonas (PTH, 1-25 dihidroxivitamina D 3 ) y por factores locales (IL-1, factor de necrosis tumoral).
El conocimiento del mecanismo molecular de la regulación de la remodelación ósea, en especial la relación entre los osteoblastos y sus células precursoras estromales con los precursores hematopoyéticos de los osteoclastos, se inició con el descubrimiento de varios miembros de la familia del TNF, como la osteoprotegerina (OPG), una proteína con una potente actividad inhibitoria de la osteoclastogénesis, seguido por el aislamiento del RANKL (un ligando trasmembrana expresado en los osteoblastos) y del RANK (un receptor de las células hematopoyéticas precursoras de los osteoclastos al que se une el RANKL).

Los radiofármacos se incorporan a la matriz ósea y al nuevo tejido óseo en formación. Durante el desarrollo embrionario los huesos largos se forman a partir de una matriz cartilaginosa, dentro de la cual surgen los núcleos de osificación primarios. La osificación endocondral va sustituyendo el tejido cartilaginoso por tejido óseo, luego aparecen en las epífisis de los huesos largos los núcleos de osificación secundarios. Esta zona de cartílago encargada del crecimiento en longitud presenta una intensa actividad metabólica hasta el desarrollo total del hueso.
El soporte vascular de las líneas de crecimiento en la zona metafisaria esta dado por las arterias metafisarias que provienen de la arteria centromedular y en las epífisis de las arterias epifisarias.




El tejido óseo es un tipo especializado de tejido conectivo constituyente principal de los huesos en los vertebrados. El tejido óseo está compuesto por células y componentes extracelulares calcificados que forman la matriz ósea. Se caracteriza por su rigidez y su gran resistencia tanto a la tracción como a la compresión. Matriz ósea: la matriz ósea representa el conjunto de la sustancia intersticial intercelular que compone el tejido óseo. Consta de: • la matriz orgánica (35%), compuesta por fibras de colágeno incluidas en una sustancia fundamental de naturaleza glicoproteica que proporcionan flexibilidad y resistencia al tejido • los componentes minerales inorgánicos (65% del peso seco del hueso) que se depositan entre la matriz orgánica, fundamentalmente fosfato cálcico que confieren la solidez característica. La dureza del hueso depende de sus componentes inorgánicos, mientras que su resistencia y elasticidad son función de la matriz orgánica, particularmente del colágeno. La matriz ósea está recorrida por un sistema de cavidades que se comunican entre sí, las células óseas se disponen en el interior o en las orillas de dichas cavidades, desde donde desempeñan su función de renovación y reabsorción de la propia matriz. Estos componentes, en asociación con las células del hueso, están altamente organizados con una histoarquitectura muy particular: la osteona o sistema de Havers. Sustancia fundamental: representa una pequeña fracción de la matriz orgánica del hueso. Compuesta por glucosaminoglucanos como el condroitín-sulfato, queratán-sulfato y ácido hialurónico. También contiene proteoglucanos de pequeño tamaño (menor al del tejido cartilaginoso) Además de estos componentes, la sustancia fundamental del tejido óseo contiene tres proteínas, no colágenas, dependientes de la vitamina D y exclusivas del hueso, que son: - Osteocalcina (2% de la matriz orgánica) - Osteopontina - Sialoproteína ósea (BSP). La sustancia fundamental contiene colágeno que constituye aproximadamente el 90% de la matriz orgánica. Mineral óseo: la sustancia inorgánica está representada por depósitos submicroscópicos de un tipo de fosfato cálcico, muy parecido al mineral hidroxiapatita: Ca10(PO4)6, pero no idéntico. Este mineral está presente en forma de pequeños cristales, como plaquitas o palillos, los cristales no tienen una distribución azarosa, sino que están situados sobre y dentro de las fibras colágenas, apareciendo regular y periódicamente a intervalos a lo largo de las fibras siguiendo la trama molecular del colágeno. También existe la presencia de varios iones: anión citrato, anión carbonato, anión fluoruro ( F −), catión sodio y catión magnesio. Pueden encontrarse cationes extraños: Plomo (Pb + + ), Estroncio (Sr + + ), y Radio (Ra + + ); e incluso isótopos radiactivos como el Estroncio (90Sr), el más peligroso junto con el Plutonio (239Pu) ya que tienen especial afinidad por el tejido óseo. Células del hueso: este tejido se renueva y se reabsorbe continuamente, gracias a la actividad de sus células específicas. Éstas son: a- los osteoblastos, responsables de la formación de tejido óseo nuevo b- los osteocitos, que son los osteoblastos maduros y desarrollan una actividad menor; c- los osteoclastos, que se encargan de reabsorber o eliminar la materia ósea. Tipos de tejido óseoMacroscópicamente se distinguen dos zonas óseas con características diferentes y sin un límite neto, éstas representan dos formas diferentes de estructuración del tejido óseo: • el hueso esponjoso: está formado por espacios vacíos o tabiques. Es un tejido reticular, tiene forma de red y entre las cavidades se encuentra la médula ósea y está recubierta por un tejido compacto. • el hueso compacto: Sus componentes están muy fusionados y es lo que le da el aspecto duro y uniforme al hueso, son abundantes en huesos largos como el fémur y el húmero.   Funciones del tejido óseo Entre las funciones del tejido óseo se encuentra la de sostener el resto del organismo, la de darle forma, la de proteger a los órganos internos y la de colaborar con los movimientos. TEMA III CRECIMIENTO Y MANTENIMIENTO DEL TEJIDO ÓSEO OSIFICACION (DESARROLLO DEL HUESO) Se denomina osificación al conjunto de mecanismos por medio de los cuales el tejido conjuntivo se transforma en tejido óseo. Mecanismos de osificación:
Tipos de osificación:
En la osificación intramembranosa se forma el hueso por diferenciación de celulas mesenquimaticas en osteoclastos. Etapa inicial de osificacion intramembranosa (craneo de embrión de gato), (Ob) osteoblastos 
HUESO INMADURO (IB) Y MADURO (MB) coloreado con hematoxilina eosina (H-E). El hueso inmaduro tiene mas celulas.  Algunos osteoblastos y fibrillas dan origen al deposito de nuevas laminillas de oseina (sustancia cementante amorfa, de por sí bastante rígida, secretada por osteoblastos) y a la vez se van cargando de sales de calcio. El tejido conjuntivo continua la multiplicación de los fibroblastos y sigue formando oseina, y el pseudo epitelio (serie de osteoblastos continua) continua retrocediendo. Así, capa a capa, va formándose el tejido óseo. Según analisis especializados o quimico común se pueden detectar las distintas sales que intervienen en el proceso tales como: carbonatos, citratos, calcio y fosfatos, por analisis químico comun; y cristales de apatita (hidroxiapatita y carbonoapatita), y cristales de citratos de calcio. Las laminillas se forman a partir de un tejido que posee sustancia fundamental, fibrillas, células conjuntivas. Durante esta formación surgen dos etapas que son la aparición de sustancia cementante (oseina) y la impregnación con sales cálcicas. Luego se genera la primera laminilla de hueso sin calcificar, luego se forma la segunda laminilla y se calcifica la primera, y asi sucesivamente. Deposito de sales cálcicas. A partir de la glucosa-6-fostato, llega a la zona de osificación y se desdobla por la fosfatasa alcalina (forfolilasa) en ion fosfato y glucosa. El ión fosfato, que es el de interes en el proceso, se une al ión calcio que proviene de la sangre y forma una sal: fosfato de calcio (83-88%). Una vez llega a su formación óptima, precipita en forma de cristales (hidroxiapatita)  
GLUCOCORTICOIDES.. Hormona segregada por la zona fasciculada de la corteza suprarrenal, como el cortisol, la cortisona y la corticosterona. Su secreción es estimulada por la hormona adenocorticoticotropa (ACTH) hipofisaria. Todas ellas aumentan el nivel de glucosa en la sangre, movilizan el calcio óseo y se comportan como antiinflamatorios. Su secreción es una de las respuestas del organismo ante el estrés. Los glucocorticoides son hormonas de acción contraria a la de la insulina en sangre. También actúan sobre el metabolismo intermedio de grasas y proteínas. Los glucocorticoides producidos por el cuerpo humano son el cortisol, la cortisona y la corticosterona. El cortisol es con diferencia el glucocorticoide más importante en el hombre. Desde el punto de vista farmacológico son corticosteroides para uso sistémico cuyo fármaco de referencia es la cortisona, regulada por la hormona hipofisaria ACTH. El cortisol y la corticosterona son secretados por las células de las capas fascicular y reticular de la corteza suprarrenal. Como son hormonas esteroides, se sintetizan a partir del colesterol, por medio de enzimas de la familia del citocromo P450, localizadas en el retículo endoplásmico liso y las mitocondrias. La actividad de algunas de estos enzimas, como el colesterol desmolasa, se incrementa por ACTH. El ACTH (corticotropina) es liberado por la hipófisis anterior en respuesta al estrés, y es el principal estimulador de la síntesis de glucocorticoides. El cortisol puede sufrir la acción del enzima 11βhidroxiesteroides deshidrogenasa, en el hígado y otros tejidos periféricos, para dar cortisona. Funciones de los glucocorticoidesEn general, se dice que los glucocorticoides son necesarios para que el organismo resista situaciones de estrés. El término "estrés" hace aquí referencia a una amplia gama de situaciones que tienen en común el hecho de que favorecen la secreción de ACTH y glucocorticoides, entre las que podríamos incluir el ayuno, la hipoglucemia, las lesiones físicas, o la ansiedad y el miedo. Los estímulos estresantes producen la muerte en los organismos que han sido adrenalectomizados. Los glucocorticoides son hormonas catabólicas. Estimulan la gluconeogénesis en hígado y riñón, de manera que elevan la glucemia. Tienen un cierto efecto antiinsulínico en muchos tejidos periféricos, lo que colabora también a aumentar la glucemia. Favorecen la degradación de proteínas y aumentan por tanto la liberación de aminoácidos a la sangre, muchos de los cuales son utilizados como sustrato de la gluconeogénesis. También tienen un efecto lipolítico. Los glucocorticoides tienen un efecto permisivo sobre otras hormonas, de manera que favorecen su función. En este sentido, facilitan los efectos termogénicos y catabólicos de catecolaminas y hormonas tiroideas. También facilitan el efecto vasopresor de las catecolaminas. El cortisol y los demás glucocorticoides tienen efecto sobre el sistema nervioso central. En la hipersecreción de corticoides (síndrome de Cushing), se produce un cierto estado de euforia, y puede llegar a producirse un trastorno psicótico franco. Los glucocorticoides tienen la capacidad de reducir dramáticamente las manifestaciones de la inflamación. Esto se debe a sus efectos considerables sobre la concentración, distribución y función de los leucocitos periféricos. Después de una sola dosis de un glucocorticoide de acción corta, la concentración de neutrófilos aumenta, mientras que los linfocitos, monocitos, eosinófilos y basófilos en la circulación disminuyen. La capacidad de estas células para responder a los antígenos y los mitógenos disminuye. Los cambios son máximos a las 6 horas y desaparecen en 24 horas. Adicionalmente, los glucocorticoides disminuyen la respuesta inflamatoria que sucede por la activación de la fosfolipasa A2. Aumentan algunos fosfolípidos que reducen la síntesis de prostaglandinas yleucotrienos. También aumentan la concentración de lipocortinas que disminuyen la disponibilidad de sustratos para la fosfolipasa. Además disminuyen la expresión de la isoforma de la ciclooxigenasa 2 (COX II). Funciones y efectos del cortisolEfectos del cortisol sobre el metabolismo de los hidratos de carbono• Estimulación de la gluconeogénesis: Es una de las funciones más conocidas del cortisol, estimula la formación de hidratos de carbono en el hígado aumentando, en la mayoría de las ocasiones, la velocidad de síntesis hasta unas seis o diez veces. Para ello el cortisol aumenta las enzimas necesarias para convertir los aminoácidos en glucosa de las células hepáticas; esto se debe al efecto de los glucocorticoides sobre la transcripción de ADN en los núcleos de las células hepáticas, que lo pasará a ARNm, y éste a su vez darán las enzimas necesarias para la glocuneogénesis. Por otro lado, el cortisol provoca la movilización de los aminoácidos para que se dispongan de más aminoácidos en el plasma para entrar en la célula hepática y promover la gluconeoneogénesis. Todo esto llevará a un aumento del glucógeno en las células hepáticas • Disminución de la utilización de la glucosa: el cortisol produce este efecto en las células de todo el organismo. Aunque se desconoce la causa, la idea más defendida es que el cortisol retrasa directamente la utilización de glucosa en algún punto entre la entrada de la glucosa a la célula y el final de su degradación. Esta idea se defiende por las observaciones en las que se comprueba que los glucocorticoides deprimen la oxidación de NADH a NAD; dado que el NADH debe oxidarse en la glucogenesis, este efecto provocaría la menor utilización de glucosa por las células • Glicemia elevada y diabetes suprarrenal: Debido a los efectos del cortisol sobre el aumento de glucógeno y la reducción moderada en la utilización de glucosa, el nivel de glicemia en sangre se ve elevado. Este proceso, si supera el 50% o más del nivel de glicemia normal se denomina diabetes suprarrenal, que tiene muchas similitudes con la diabetes hipofisaria; Por otro lado, en la diabetes suprarrenal, la insulina reduce moderadamente estos niveles, mientras que en la hepática la reducción es mucho mayor y es mayor que en la hipofisaria. Efectos del cortisol sobre el metabolismo de las proteínas• Reducción de las proteínas celulares. El cortisol produce estos efectos en todas las células, menos en las hepáticas. Provoca una disminución de la síntesis de proteínas y un aumento del catabolismo de las mismas; esto se da debido a que el cortisol deprime la formación de ARN y a una disminución del transporte de aminoácidos (sobre todo se da en el músculo y en el tejido linfoide. Si existe mucho cortisol el músculo puede llegar a debilitarse tanto que no sea capaz de mantenerse de pie; y las funciones linfocitarias pueden reducirse hasta una pequeña fracción de lo normal. • Aumento de las proteínas hepáticas. Al disminuir las proteínas de todo el organismo, las de las células hepáticas aumentan (también las de las células plasmáticas producidas por el hígado. La diferencia de producción entre las células extrahepáticas y las hepáticas, se cree que es debido a un efecto de cortisol que favorece el transporte de aminoácidos hacia el interior de las células hepáticas (y no a las otras células) y de las enzimas hepáticas necesarias para la síntesis de proteínas. • Aumento de los aminoácidos sanguíneos, disminución del transporte de aminoácidos a las células extrahepáticas y aumento del transporte de células hepáticas. Se ha comprobado que el cortisol deprime el transporte de aminoácidos al interior de las células musculares y, quizás de otras células exrahepáticas. La disminución del transporte de aminoácidos al interior de las células extrahepáticas, reduce la concentración interna de aminoácidos, y en consecuencia, disminuye la síntesis de proteínas. Sin embargo, el catabolismo proteico de las células, continuará enviando aminoácidos (de las proteínas ya existentes) al medio intracelular, para aumentar allí su concentración. Por lo tanto, el cortisol moviliza los aminoácidos de los tejidos no hepáticos, con lo que disminuirán los depósitos tisulares de proteínas. El aumento de la concentración de aminoácidos, y que el cortisol favorece su transporte al interior de las células hepáticas podría explicar los efectos del hígado para producir: a) Aumento de la tasa de desaminación de aminoácidos b) Aumento de la síntesis de proteínas por el hígado c) Aumento de la formación de proteínas plasmáticas d) Aumento de la conversión de aminoácidos en glucosa (aumento de la neoglucogénesis Por tanto, es posible que muchos efectos del cortisol sobre los sistemas metabólicos del organismo sean consecuencia de su capacidad para movilizar aminoácidos de los tejidos periféricos al tiempo que aumenta las enzimas hepáticas necesarias para lograr los efectos hepáticos Efectos del cortisol sobre el metabolismo de las grasas• Movilización de ácidos grasos. De una forma similar a la movilización de aminoácidos del músculo, el cortisol va a promover la movilización de ácidos grasos del tejido adiposo. Esta movilizacón dará como resultado un aumento de la concentración de los ácidos grasos en el plasma. Este transporte se cree que es promovido por la disminución del transporte de glucosa a los adipocitos, que suele ocurrir en condiciones extremas (como la falta de alimentación),donde será necesaria otra fuente de energía (los ácidos grasos). Se trata de un mecanismo lento, que requiere de varias horas. • Obesidad causada por el cortisol. Aquelllas personas que presenten una secrección de cortisol excesiva, presentarán una obesidad caracterizada por un deposito excesivo de grasa en el tórax, y en las regiones craneales. Se cree que se produce como sonsecuencia de una sobrealimentació Función del cortisol en el estrés y en la inflamaciónLos traumatismos, infecciones, calor o frío intenso, inyecciones de noradrenalina, cirugías, inyecciones subcutáneas de necrotizantes y cualquier enfermedad debilitante provocan un aumento de la liberación de cortisol. Se cree que este aumento puede ser beneficioso porque movilizan rápidamente las grasas y aminoácidos(esto facilita la producción de energía y la formación de sustancias como la glucosa,purinas o pirimidinas). • Efectos antiinflamatorios del cortisol La principal función del cortisol es la de bloquear la inflamación causada por cualquier tipo de daño, e incluso anularla antes de que ocurra (véase el proceso de inflamación) La inflamación es inhibida por parte del cortisol, en las primeras etapas (donde se libera gran cantidad de sustancias que activan el proceso inflamatorio y aumenta el flujo sanguíeo y la salida de plasma en la zona dañada)o incluso antes, y si ha comenzado, la finaliza rápidamente. Uno de los efectos antiinflamatorios más importantes que provoca el cortisol, es la estabilización de las membranas de los lisosomas; hace más resistentes a estas membranas, evitando así que se libere el contenido enzimático que contienen y que aumenta la inflamación. Otro de los efectos, es la impermeabilidad de los vasos; los hace menos permeables para que se libere menos plasma. Un tercer efecto, es que disminuye la formación de prostaglandinas y leucotrienos, lo que provoca una reducción de la movilización de leucocitos y de la fagocitosis en la zona afectada Además, reduce la producción de linfocitos T porque inhibe el sistema inmunitario (esto reduce las reacciones que se producen en el tejido y que aumentarían la inflamación). Y por último, reduce la temperatura (evita la fiebre) debido a que reduce los niveles de interleuquina-I, y como consecuencia también se reduce la vasodilatación. Incluso cuando la inflamación ha madurado, la administración de cortisol provoca una reducción de los efectos en poco tiempo(quizás como consecuencia de la movilización de aminoácidos para reparar los tejidos, por el aumento en la producción de glucosa, o por la movilización de grasas para aporte energético). Por todo lo mencionado, la administración de cortisol, es importante en el tratamiento de enfermedades como la artritis reumatoide, la fiebre reumática o la glomerulo nefritis. Aunque el cortisol no corrige el proceso básico de la inflamación, pero evita los efectos nocivos de su respuesta.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
¿Qué es el estroncio? |
El estroncio es un elemento natural que se encuentra en rocas, el suelo, polvo, carbón y petróleo. El estroncio natural no es radioactivo y se conoce como estroncio estable o simplemente estroncio. En el ambiente existen cuatro isótopos del estroncio estable, 84Sr, 86Sr, 87Sr y 88Sr (léase estroncio ochenta y cuatro, etc.). Los compuestos de estroncio se usan en la fabricación de cerámicas y productos de vidrio, fuegos artificiales, pigmentos para pinturas, luces fluorescentes y medicamentos. El estroncio puede existir también en forma de varios isótopos radioactivos, el más común es el 90Sr. El 90Sr se forma en reactores nucleares o durante la detonación de armas nucleares. El estroncio radioactivo genera partículas beta a medida que decae. Una de las propiedades radioactivas del estroncio es la vida-media, o el tiempo que toma la mitad del isótopo en emitir su radiación y transformarse en otra sustancia. La vida-media del 90Sr es 29 años. |
¿Qué le sucede al estroncio cuando entra al medio ambiente? |
|
¿Cómo podría yo estar expuesto al estroncio? |
|
¿Cómo puede afectar mi salud el estroncio? |
No se ha demostrado que la exposición a niveles bajos de estroncio estable afecta la salud de adultos, pero puede perjudicar a los niños (vea la sección que sigue). No se ha demostrado que respirar o ingerir niveles bajos niveles de estroncio radioactivo perjudica la salud. Los niveles altos de estroncio radioactivo pueden dañar la médula de los huesos y producir anemia y prevenir que la sangre coagule apropiadamente. |
¿Qué posibilidades hay de que el estroncio produzca cáncer? |
| El único compuesto de estroncio estable que puede producir cáncer es el cromato de estroncio, pero esto se debe al cromo y no al estroncio. La exposición a niveles altos de estroncio radioactivo puede producir cáncer. En seres humanos expuestos a cantidades relativamente altas de estroncio se ha observado leucemia. En animales de laboratorio también se han observado leucemia y cáncer de los huesos, la nariz, los pulmones y la piel. La Agencia Internacional para la Investigación del Cáncer (IARC) ha determinado que el estroncio radioactivo es carcinogénico en seres humanos. |
¿Cómo puede el estroncio afectar a los niños? |
No sabemos si la exposición al estroncio producirá defectos de nacimiento u otros efectos sobre el desarrollo en seres humanos. En animales expuestos a estroncio radioactivo se han observado defectos de nacimiento. La exposición a niveles altos de estroncio estable puede alterar el crecimiento de los huesos en los niños. Los niños pueden ser más susceptibles que los adultos a los efectos perjudiciales del estroncio radioactivo. |
¿Cómo pueden las familias reducir el riesgo de exposición al estroncio? |
Mantener una dieta balanceada con suficientes cantidades de vitamina D, calcio y proteínas reducirá la cantidad de estroncio que es absorbida. |
¿Hay algún examen médico que demuestre que he estado expuesto al estroncio? |
Toda la gente tiene pequeñas cantidades de estroncio estable en el cuerpo. Hay exámenes para medir el nivel de estroncio en la sangre, el cabello, las heces y la orina. Estos exámenes son de mayor utilidad para gente expuesta a niveles altos de estroncio. Estos exámenes no pueden determinar los niveles exactos de estroncio a los que puede haber estado expuesto o predecir la manera en que estos niveles afectarán su salud. Hay dos tipos de pruebas disponibles para el estroncio radioactivo. En la primera se determina si usted ha estado expuesto a una dosis alta de radiación; en la segunda se determina si hay estroncio en su cuerpo. En la primera prueba se evalúan cambios en el número de células en la sangre o en los cromosomas que ocurren a niveles de exposición 3 a 5 veces más altos que el límite de la dosis ocupacional anual. Esta prueba no puede indicar si la radiación provino del estroncio. En el segundo tipo de prueba se examinan la sangre, las heces, la saliva, la orina, los dientes o el cuerpo entero. El propósito es determinar si el estroncio está siendo eliminado del cuerpo, si se encuentra en sus dientes o si es retenido en el cuerpo en niveles más altos que lo normal. Las muestras pueden ser tomadas en el consultorio del doctor para enviarse a un laboratorio especial o usted debe ir directamente al laboratorio. FlúorSímbolo F, número atómico 9, miembro de la familia de los halógenos con el número y peso atómicos más bajos. Aunque sólo el isótopo con peso atómico 19 es estable, se han preparado de manera artificial los isótopos radiactivos, con pesos atómicos 17 y 22, el flúor es el elemento más electronegativo, y por un margen importante, el elemento no metálico más energético químicamente. Propiedades: El flúor elemental es un gas de color amarillo pálido a temperaturas normales. El olor del elemento es algo que está todavía en duda. La reactividad del elemento es tan grande que reacciona con facilidad, a temperatura ambiente, con muchas otras sustancias elementales, entre ellas el azufre, el yodo, el fósforo, el bromo y la mayor parte de los metales. Dado que los productos de reacción con los no metales son líquidos o gases, las reacciones continúan hasta consumirlo por completo, con frecuencia con producción considerable de calor y luz. En las reacciones con los metales forma un fluoruro metálico protector que bloquea una reacción posterior a menos que la temperatura se eleve. El aluminio, el níquel, el magnesio y el cobre forman tales películas de fluoruro protector. El flúor reacciona con violencia considerable con la mayor parte de los compuestos que contienen hidrógeno, como el agua, el amoniaco y todas las sustancias orgánicas, sean líquidos, sólidos o gases. La reacción del flúor con el agua es compleja y produce principalmente fluoruro de hidrógeno y oxígeno, así como cantidades menores de peróxido de hidrógeno, difluoruro de oxígeno y ozono. El flúor desplaza otros elementos no metálicos de sus compuestos, aun aquellos muy cercanos en cuanto a actividad química. Desplaza el cloro del cloruro de sodio y el oxígeno en la sílica, en vidrio y en algunos materiales cerámicos. En ausencia de fluoruro de hidrógeno, el flúor no ataca en forma significativa al cuarzo o al vidrio, ni aun después de varias horas a temperaturas hasta de 200ºC (390ºF). El flúor es un elemento muy tóxico y reactivo. Muchos de sus compuestos, en especial los inorgánicos, son también tóxicos y pueden causar quemaduras severas y profundas. Hay que tener cuidado para prevenir que líquidos o vapores entren en contacto con la piel y los ojos. Frecuencia natural: Se estima que se halla en un 0.065% en la corteza terrestre; es casi tan abundante como el carbono, el nitrógeno o el cloro, mucho más que el cobre o el plomo, aunque mucho menos que el hierro, aluminio o el magnesio. Los compuestos cuyas moléculas contienen átomos de flúor están ampliamente distribuidos en la naturaleza. Muchos minerales contienen cantidades pequeñas del elemento, y se encuentra tanto en rocas ígneas como en rocas sedimentarias. Aplicaciones: Los compuestos que contienen flúor se utilizan para incrementar la fluidez del vidrio fundido y escorias en la industria vidriera y cerámica. El espato flúor (fluoruro de calcio) se introduce dentro del alto horno para reducir la viscosidad de la escoria en la metalurgia del hierro. La criolita, Na2AlF6, se utiliza para formar el electrólito en la metalurgia del aluminio. El óxido de aluminio se disuelve en este electrólito, y el metal se reduce, eléctricamente, de la masa fundida. El uso de halocarburos que contienen flúor como refrigerantes se patentó en 1930, y estos compuestos estables y volátiles encontraron un mercado como propelentes de aerosoles, así como también en refrigeración y en sistemas de aire acondicionado. Sin embargo, el empleo de fluorocarburos como propelentes ha disminuido en forma considerable a causa del posible daño; a la capa de ozono de la atmósfera. Un uso del flúor, muy importante durante la Segunda Guerra Mundial, fue un el enriquecimiento del isótopo fisionable 235U; el proceso más importante empleaba hexafluoruro de uranio. Este compuesto estable y volátil fue con mucho el material más adecuado para la separación del isótopo por difusión gaseosa. Mientras que para los consumidores la utilización de compuestos de flúor en la industria pasa casi inadvertida, algunos compuestos se han vuelto familiares a través de usos menores pero importantes, como aditivos en pastas de dientes y superficies fluoropoliméricas antiadherentes sobre sartenes y hojas de afeitar (teflón por ejemplo). Compuestos: En todos los compuestos de flúor la alta electronegatividad de este compuesto indica que el átomo de flúor tiene un exceso de carga negativa. Es conveniente, sin embargo, dividir los fluoruros binarios inorgánicos en sales (red iónica), fluoruros metálicos no volátiles y fluoruros volátiles, la mayor parte de no metales. Algunos hexafluoruros metálicos y los fluoruros de gases nobles muestran volatilidad que son frecuencia está asociada a un compuesto molecular. La volatilidad se asocia a menudo con números de oxidación altos para el elemento positivo. Los metales suelen formar fluoruros iónicos no volátiles, donde la transferencia electrónica es sustancial y la red cristalina está determinada por el tamaño iónico y la interacción electrostática predecible. Cuando el número de coordinación y la valencia son la misma, por ejemplo en BF3, SiF4 y WF6, el enlace entre el metal y el flúor no es común; los compuestos resultantes son muy volátiles y los sólidos muestran redes moleculares más que estructuras cristalinas iónicas. Para números de oxidación superiores, las redes iónicas simples son menos comunes y, mientras que el enlace entre el átomo central y el flúor requiere aún transferencia de alguna carga al flúor, las estructuras moleculares son identificables en las fases condensadas. Además de los fluoruros binarios, se ha aislado un número muy grande de complejos, a menudo con un anión fluoruro que contiene un átomo central de número de oxidación alto. Los fluoruros binarios salinos muestran una gran tendencia a combinarse con otros fluoruros binarios para formar numerosos complejos o sales dobles. Los compuestos de carbono que contienen flúor pueden dividirse en hidrocarburos fluorados y derivados (compuestos orgánicos del flúor), y los fluorocarburos y sus derivados. El átomo de flúor unido al anillo aromático, como en el fluorobenceno, es poco reactivo. Además reduce la reactividad de toda la molécula. Por ejemplo, aquellos colorantes que contienen flúor unido al anillo aromático son más resistentes a la oxidación y más sensibles a la luz, que los que no lo contienen. La mayor parte de los compuestos alifáticos, como los fluoruros de alquilo, son inestables y pierden fluoruro de hidrógeno con facilidad. Estos compuestos son difíciles de preparar y conservar, y es poco probable que se vuelvan importantes. Efectos del Flúor sobre la saludEn el agua, aire, plantas y animales hay presentes pequeñas cantidades de flúor. Como resultado los humanos están expuestos al flúor a través de los alimentos y el agua potable y al respirar el aire. El flúor se puede encontrar en cualquier tipo de comida en cantidades relativamente pequeñas. Se pueden encontrar grandes cantidades de flúor en el té y en los mariscos. El flúor es esencial para mantener la solidez de nuestros huesos. El flúor también nos puede proteger del decaimiento dental, si es aplicado con el dentifríco dos veces al día. Si se absorbe flúor con demasiada frecuencia, puede provocar caries, osteoporosis y daños a los riñones, huesos, nervios y músculos. Las industrias liberan la forma gaseosa del flúor. Este gas es muy peligroso, ya que en elevadas concentraciones puede causar la muerte. En bajas concentraciones puede causar irritaciones de los ojos y la nariz. Efectos ambientales del FlúorEl flúor está presente en la corteza terrestre de forma natural, pudiendo ser encontrado en rocas, carbón y arcilla. Los fluoruros son liberados al aire cuando el viento arrastra el suelo. Los procesos de combustión en las industrias pueden liberar fluoruro de hidrógeno al aire. Los fluoruros que se encuentran en el aire acabarán depositándose en el suelo o en el agua. Cuando el flúor se fija a partículas muy pequeñas puede permanecer en el aire durante un largo periodo de tiempo. Cuando el flúor del aire acaba en el agua se instala en los sedimentos. Cuando acaba en los suelos, el flúor se pega fuertemente a las partículas del suelo. En el medio ambiente el flúor no puede ser destruído; solamente puede cambiar de forma. El flúor que se encuentra en el suelo puede acumularse en las plantas. La cantidad de flúor que tomen las plantas depende del tipo de planta, del tipo de suelo y de la cantidad y tipo de flúor que se encuentre en el suelo. En las plantas que son sensibles a la exposición del flúor incluso bajas concentraciones de flúor pueden provocar daños en las hojas y una disminución del crecimiento. Los animales que ingieren plantas que contienen flúor pueden acumular grandes cantidades de flúor en sus cuerpos. El flúor se acumula principalmente en los huesos. Como consecuencia, los animales expuestos a elevadas concentraciones de flúor sufren de caries y degradación de los huesos. Demasiado flúor también puede provocar la disminución de la cantidad de alimento tomado por el estómago y puede alterar el desarrollo de las garras. Por último, puede provocar bajo peso al nacer. Calcio, Fósforo, Vitamina D y Parathormona.) METABOLISMO DEL CALCIO Y FÓSFORO INORGÁNICO Más del 70% de las cenizas del cuerpo consisten en Ca y PO4. Más del 99% del total de Ca, y 80-85% del P están contenidos en el esqueleto y muelas. La menor proporción presente en los fluidos corporales juega un rol extremadamente importante en el mantenimiento de las funciones corporales. El Ca en el fluido extracelular es crítico para: • normal excitabilidad neuromuscular • permeabilidad capilar y de las membranas celulares • contracción muscular • transmisión normal del impulso nervioso • coagulación sanguínea normal El fósforo fuera del esqueleto juega a menudo un rol fascinante. El mismo está comprometido en la estructura celular normal y sirve en la degradación y síntesis de numerosos compuestos de carbono. En las uniones de alta energía juega un rol fundamental en el depósito, liberación y transferencia de energía. Finalmente, la habilidad del fósforo para ser excretado como H2PO4- o HPO42, proporciona un margen amplio para regular el metabolismo ácido-base. A - CALCIO Y FÓSFORO EN COMPOSICIÓN Y FORMACIÓN DEL HUESO El hueso adulto normal está compuesto por 45% de agua, 25% de cenizas, 20% de proteínas y 10% de grasas. En el mamífero las cenizas están compuestas por 36% de Ca, 17% de PO4 y 0.8% de Mg. El calcio se encuentra en el hueso en parte como fosfato tricálcico y parte como carbonato, en una compleja estructura de apatita (hidroxiapatita) y cuya fórmula se escribe CaCO3xnCa3(PO4)2. Sin embargo, el hueso no parece tener una composición constante. Los cristales aparentemente absorben y admiten varios grupos de iones sin carga con una geometría de cristales enrejados. De aquí que el hueso contenga, a parte de Ca y PO4, proporciones variables de carbonato, fluoruro, citrato, N+, K+, y Mg++. Hay una clara diferencia entre el hueso de un animal joven y uno adulto, la sal que determina la solubilidad en el hueso joven es el fosfato octocálcico, y en el adulto el hueso está formado por hidroxiapatita. Esta diferencia explica la alta concentración de fósforo inorgánico (Pi) en animales jóvenes. Los minerales del hueso están dispuestos para ser movilizados rápidamente y mantener el nivel de Ca sérico, pero menos lo están para mantener el nivel de fósforo, de forma tal que una disminución en el nivel de fósforo sérico es el primer signo de la deficiencia de consumo de este mineral. Osificación: Fisiología Ósea El hueso está formado por los osteoblastos, células que sintetizan la matriz proteica primero, y posteriormente regulan la mineralización primaria responsable del 75-85 % de la mineralización total del hueso. Los osteoclastos los cuales efectúan la resorción ósea, proceso en el cual se remueve simultáneamente la fase mineral y se digiere la porción orgánica del hueso. Los osteoblastos activos cubren el 5% de la superficie ósea, el 10% lo ocupan los osteoblastos en reposo. El 80% restante de la superficie ósea está cubierta por unas células aplanadas, los osteocitos superficiales. Los osteoblastos que quedan incluidos en el tejido óseo se denominan osteocitos profundos, y forman a su alrededor un tejido óseo metabólicamente muy activo y que pueden variar rápidamente su contenido mineral en respuesta a las necesidades. En el hueso coexisten dos procesos: A) Modelamiento óseo, que incluye el crecimiento longitudinal que se detiene después de la pubertad y las modificaciones del diámetro transversal que son continuas y se efectúan por aposición perióstica y por resorción endostal. En este caso la formación y la resorción ocurren en diferentes superficies óseas. B) Remodelamiento óseo, que ocurre en el hueso trabecular y cortical. Este proceso ocurre de la siguiente manera: un grupo de osteoclastos excava el hueso cortical longitudinalmente, este frente osteoclástico avanza en forma centrífuga (resorción), lo sigue una zona de transición y luego una capa de osteoblastos avanzando en sentido centrípeto forma tejido óseo que va cerrando radialmente el túnel excavado (deposición). Este sistema completo se denomina Unidad Cortical de Remodelamiento Óseo (UCRO). Intercambio de Ca entre el líquido extracelular óseo y el sistémico El líquido extracelular óseo (LECO) está en contacto con los cristales óseos de los canalículos, lagunas y entre los osteocitos de superficie y tejido óseo. Tiene una composición iónica diferente al líquido extracelular sistémico (LECS). Los osteocitos profundos y los de superficie facilitan el pasaje del Ca iónico del líquido extracelular óseo al sistémico. Este fenómeno resulta de la producción de un diferente gradiente de concentración entre sangre y hueso. El Ca pasa continuamente hacia el LEC óseo por diferencia en el gradiente de concentración, y sería a su vez removido activamente por los osteocitos. La magnitud de este flujo depende de la diferencia de concentración generado por los osteocitos. Este sistema de intercambio de Ca entre el LEC óseo y el sistémico permite mantener una concentración de Ca plasmático de aproximadamente 6 mg/% aún en ausencia completa de PTH. B - ABSORCIÓN DE CALCIO Y FÓSFORO El Ca contenido en la dieta per se determinaría la cantidad absorbida desde el tracto gastrointestinal, y el nivel de Ca sérico tiende a aumentar o disminuir con el nivel de Ca en la dieta. Esto es verdad en ovinos, pero en la vaca es difícil provocar una disminución de Ca por ingerir una dieta deficiente en Ca. Pero dietas con relación Ca/PO4 elevadas pueden causar un leve aumento en el nivel de Ca sérico. La eficiencia de la absorción de Ca puede aumentar en condiciones que causan aumentos de la demanda (preñez y lactancia). La absorción de Ca es pasiva en parte “adaptativa”, es decir, en crisis hipocalcémicas se pone en juego una secuencia de eventos desencadenados por la acción de la Vitamina D activa o 17 OH – Colecalciferol la cual eleva la síntesis a nivel intestinal de una proteína transportadora de Ca denominada PB-Ca esta toma el elemento de la luz intestinal volcándola al torrente circulatorio. La formación de compuestos insolubles a nivel intestinal reduce la absorción de Ca. Se demostró que el oxalato puede reducir la absorción del mineral formando oxalato de Ca insoluble que se pierde en heces. En rumiantes el oxalato se descompone por acción microbiana. La degradación del oxalato puede resultar en alcalosis, entonces, indirectamente afecta el metabolismo del Ca. Algunos subproductos como la remolacha forrajera y la alfalfa en ciertos momentos de maduración contiene más del 7% de oxalato de Ca. Esto puede alterar la capacidad del rúmen para metabolizar el oxalato, inmoviliza el Ca en el tracto digestivo y contribuye a desarrollar una hipocalcemia. En monogástricos los fitatos presentes en altas concentraciones en cereales tienen una considerable importancia en el metabolismo del Ca y del PO4. El PO4 unido de esta manera no es movilizable por los monogástricos ya que estos no contienen las hidrolizas específicas denominadas fitasas intestinales. Si esto no ocurre el cuerpo no sólo es despojado de PO4, sino también de Ca. Esto es debido a que las fitasas(Acido Inositol Hexa Fosfórico) a pH 5-7 forman compuestos muy insolubles en el intestino. La velocidad de absorción del Ca depende, como ya se ha mencionado, de los requerimientos totales del organismo y la absorción es máxima durante el crecimiento. Durante los períodos de rápido crecimiento, las cantidades absorbidas son grandes pero la relación Calcio retenido/Peso ganado puede no ser óptima y entonces, el cociente Ca/Masa corporal es bajo y el resultado es un esqueleto mal calcificado. Esto puede presentarse en animales alimentados a leche de vaca con índices de crecimiento altos como cachorros de perro y cerdo. Los terneros crecen relativamente despacio, como la leche de vaca tiene baja concentración de Ca respecto a los nutrientes responsables del crecimiento la fracción Calcio retenido/ Crecimiento total es baja y puede aparecer raquitismo fundamentalmente cuando la alimentación es exclusivamente láctea. Es lógico suponer que a un pH intestinal bajo aumenta la proporción de PO4HCa soluble y se incrementaría su disponibilidad; al contrario, al aumentar el pH se forma (PO4)2Ca2 insoluble y la proporción de Ca absorbible disminuye. La acidificación aumenta la fracción urinaria de Ca (Ca ultrafiltrable). Ciertos ácidos retrasan o impiden la absorción intestinal de Ca. La presencia de AG no absorbibles en el intestino forma jabones cálcicos no absorbibles. El fosfato influye de una manera notable sobre la absorción de Ca. Es importante la relación Ca/PO4- a nivel del intestino delgado, ya que una intensa elevación del Fósforo forma fosfatos insolubles no absorbibles. El Ca es absorbido principalmente en la porción anterior del intestino delgado, donde juega un rol fundamental el pH pues cuanto mayor es la acidez, la absorción es mayor. El Fósforo se absorbe en dos sitios: rúmen e intestino delgado, aunque el rúmen es bastante impermeable para el ion fosfato. C - VITAMINA D La vitamina D es un factor muy importante en la regulación de la absorción de Ca; tiene un efecto directo sobre la mineralización del hueso, como también en la absorción intestinal de Ca. Esta vitamina está involucrada en el mecanismo que balancea el Ca esquelético y sanguíneo. La vitamina D se puede considerar una Pre-Hormona que origina una hormona renal, la 1,25 Di-Hidroxi-Vitamina D3 [1,25 (OH)2-D3]. Otro metabolito de la Vitamina D, la 24,25 Di-Hidroxi-Vitamina D3 [24,25 (OH)2-D3], podría así mismo ser otra hormona renal derivada de la vitamina D. Estructura química y síntesis Bajo la acción de los rayos ultravioletas, el precursor 7-di-hidroxi-colesterol sufre una alteración de su estructura: la apertura del anillo B del núcleo. Luego bajo la acción de la temperatura aparece otra doble ligadura, lo cual concluye con el proceso de formación de Vitamina D endógena. Rutas Metabólicas de la Vitamina D3 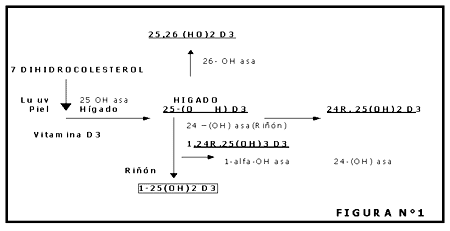 Metabolismo de la Vitamina D Sea el origen exógeno o endógeno, la Vitamina D circula en plasma y es captada rápidamente por el hígado donde sufre su primera transformación que la convierte en 25 OH-D3, la forma circulatoria predominante, incluidos la vitamina D y sus metabolitos. Esta reacción es catalizada por la enzima 25-Hidroxilasa presente en la fracción microsómica del hígado. La conversión de vitamina D a 25-HO D3 carece prácticamente de sistema de regulación, por lo que la administración de vitamina D en dosis farmacológicas produce incrementos anormales en la tasa de circulante de 25-HO-D3. La 25-OH-D3 es transportada por una alfaglobulina de Peso Molecular 52.000, el mismo sistema se utiliza para el transporte de la Vitamina D y todos sus metabolitos, pero la 25-OH-D3 tiene la mayor afinidad. Metabolismo renal de la 25-OH-D3 En concentraciones fisiológicas la 25-OH-D3 no actuaría sobre ningún proceso fisiológico. Para ello debe ser activada lo que ocurre en el riñón a nivel mitocondrial, bajo la acción de la enzima 25-OH-D3 – 1-°-hidroxilasa (1-°-Hasa). En este sistema enzimático, una flavoproteína, una proteína ferrosulfurada y el Citocromo P-450, participan en la hidroxilación del sustrato. La 1,25 (OH)2-D3 satisface todos los requerimientos para ser considerada una hormona. Se origina a partir del sistema Pre-Hormona Vitamina D - 1,25 (OH)2-D3 en un órgano específico: el riñón. La velocidad de síntesis está regulada por un complejo mecanismo en el que participan iones y hormonas, como se verá más adelante. Una vez formada la 1,25 (OH)2-D3, la misma ejerce sus efectos a nivel intestinal, renal y óseo. Otro sistema enzimático, presente en el riñón, convierte a la 25-OH-D3 en otro esteroide: la 24,25 (OH)2-D3. Laenzima que actúa en esta OH transformación es la 25-OH-D3 – 24 Hidroxilasa (24-OHasa), que se encuentra en el riñón, intestino y cartílago. La hidroxilación en la posición 24 sería el paso inicial en la inactivación del sustrato 25-OH-D3, y también en la inactivación de la 1,25 (OH)2-D3, ya que este esteroide se hidroxila asimismo en el trihidroxilado 1,24,25 (OH)3-D3. Regulación del Metabolismo de la Vitamina D El organismo carece de mecanismos de regulación de la síntesis endógena de vitamina D o de su aporte exógeno. Por lo tanto un ingreso elevado por dieta o inyección, inducen, sin mecanismos que se opongan, una elevación en la tasa circulante de la vitamina. Ya hemos mencionado antes que el primer paso en el metabolismo de la vita-mina D, la hidroxilación hepática que produce la 25-HO-D3, se produce sin control humoral u hormonal. En síntesis, la cantidad de 25-HO.D3, en el plasma es una función directa del aporte exógeno o endógeno de la vitamina. Como este aporte fluctúa a lo largo del año significativamente y de las distintas estaciones y se trata de una vitamina que regula una función orgánica de primordial importancia (el nivel de calcio en el líquido extracelular), no es extraño que 25-HO-D3 tenga una acción débil o nula sobre los tejidos efectores. La acción biológica es ejercida por los productos renales, la 1,25 (OH)2-D3 y posiblemente también, la 24,25 (OH)2-D3. Es a este nivel que se ha hallado un complejo sistema de regulación que permitiría al organismo contar con las cantidades necesarias de estas hormonas renales, y regulan así, eficazmente el metabolismo del Ca y PO4. FIGURA N° 2 Rutas Metabólicas de la Vitamina D Inactivación de la 25(HO)2D3 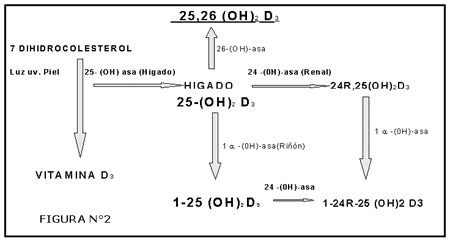 Regulación de la 1-°-HOasa renal Un aporte exógeno deficiente de Ca o niveles disminuidos de Ca circulante, aumentan la síntesis de 1,25 (OH)2-D3. El nivel de Ca controla la síntesis renal de la 1,25 (OH)2-D3 en conjunción con la Paratohormona (PTH). Esta hormona provoca un aumento en la actividad de la 1-a-OHasa renal. Otro importante factor de regulación es el fosfato. Si la concentración de PO4 aumenta considerablemente, la síntesis de 1,25 (OH)2-D3 está disminuida, aún cuando la PTH se encuentra elevada. Una disminución de la fosfatemia estimu-la la 1-a-OHasa renal y aumenta la secreción de 1,25 (OH)2-D3. Por lo tanto, la hipocalcemia estimula la síntesis de 1,25 (OH)2-D3 solo en presencia de hormona Paratiroidea. La Prolactina y los estrógenos aumentan la actividad de la 1-a-ohasa, como así también la Somatotrofina y el cortisol. Regulación de la enzima 24-HOasa renal En todos los casos en que se inhibe o estimula la síntesis de 1,25 (OH)2-D3 se observa inmediatamente un efecto inverso sobre la producción de 24,25 (OH)2-D3. Tanto la hipercalcemia, como la hiperfosfatemia, aumenta la síntesis de 24,25 (OH)2-D3. Si estos mecanismos son los que corresponden a la regulación de una hormona o son solo producto de la degradación de la 25-(HO)-D3 cuando no existen necesidades sistémicas de 1,25-(OH)2-D3 es un tema que será motivo de nuevas investigaciones, pero más adelante comentaremos el papel fisiológi-co de este metabolito. Acción biológica de la vitamina D y sus metabolitos. Este compuesto aumenta la absorción intestinal de Ca y PO4, consecuentemente aumenta la concentración sistémica y promueve la mineralización de la sustancia osteoide recién formada. A grandes rasgos se pueden mencionar dos interpretaciones: la primera sugiere que la vitamina D origina la hormona renal 1,25(HO)-D3, que sería la única encargada de producir el efecto biológico de-seado. La segunda, en cambio, sostiene que los diferentes metabolitos, de la vitamina D tendrían acciones biológicas independientes y que su interdependencia tendería a una mejor regulación del metabolismo óseo y mineral. (VER FIGURA N° 2) Efecto intestinal La 1,25 (OH)2-D3 estimula la absorción de Ca actuando en forma directa sobre las células del epitelio intestinal. El transporte de Ca estimulado por la 1,25 (OH)2-D3 se produce a través de un proceso activo que requiere gasto de energía contra un gradiente electroquímico. El sitio principal de acción es el duodeno y la primera porción del yeyuno. El tiempo entre la administración de 1,25 (OH)2-D3 y el aumento de la calcemia es de 3 a 4 horas, en contraposición a la administración de vitamina D cuyo intervalo es mayor (de 8 a 10 horas). Además, la 1,25 (OH)2-D3 estimula el transporte de fosfato. Esta acción es independiente del transporte de Ca, y se produce por un mecanismo de transporte activo dependiente del Na. El sitio principal de acción es a nivel de yeyuno e íleon. En alta dosis la 25-OH-D3 también es capaz de estimular la absorción de Ca y PO4. Efecto óseo La 1,25 (OH)2-D3 produce una elevación de la calcemia muy importante, trabajando a nivel de osteclastos, aumentando el número y actividad y estimulando la totalidad de la superficie de resorción. La 25-OH-D3 es el compuesto más potente para estimular la mineralización ósea. La 1,25 (OH)2-D3 sería en principio un metabolito sintetizado ante un estrés hipocalcémico y su papel fisiológico sería (junto con la PTH) el restablecimiento rápido de la calcemia estimulando la absorción intestinal y actuando directamente sobre el hueso para promover la resorción del tejido óseo, con liberación de Ca y otros minerales al LEC. Una vez restablecida la calcemia cesa la síntesis de 1,25 (OH)2-D3, y comienza la producción de 24,25 (OH)2-D3, facilitando esta el pasaje de Ca del LEC sistémico al LEC óseo, promoviendo la calci-ficación del hueso. La acción de la 25- (HO)- D3 se explica en principio por su conversión a 1,25 (OH)2-D3 con recuperación de la calcemia, estimulando luego activamente la mineralización del hueso por intermedio de la 24,25-(OH)2-D3. Esta teoría, ya fundamentada por experiencias recientes explicaría por una parte, el reconocido efecto de la vitamina D sobre la calcificación ósea, por vía de la síntesis de 25-(HO)-D3 y 24,25-(OH)2-D3- y por otra parte, la evidente estimulación de la resorción ósea inducida por la 1,25-(OH)2-D3 Efecto renal Tanto la 1,25 (OH)2-D3 como la 25-OH-D3 aumentan la resorción de Ca y PO4. La 1,25 (OH)2-D3 es sinérgica a la PTH con respecto al Ca y antagónica con respecto al PO4. La administración de vitamina D eleva la calcemia, ésta disminuye la concentración de Paratohormona, y de esta forma se reduce la acción de esta hormona sobre el riñón con aumento de la calciuria y disminución de la fosfaturia. Por lo tanto la vitamina D actúa sobre la regulación renal del calcio por dos vías: 1. facilitando su reabsorción 2. incrementando la respuesta del túbulo a la PTH La administración de vitamina D induce la síntesis de una proteína ligadora del calcio en el túbulo contorneado distal y en el túbulo colector, puntos de acción de la PHT. En el riñón, la 1,25-(OH)2-D3 inhibe la actividad de la 25-HO-D-1a-hidroxilasa, y estimula la actividad de la 25-HO-D-24-hiroxilasa, con la consiguiente forma-ción de 24,25-(OH)2-D3. Mecanismo de acción de 1,25 (OH)2-D3 Como otras hormonas esteroides, la 1,25 (OH)2-D3 actúa a través de la estimulación nuclear de la síntesis de ARNm y la inducción de proteínas que influyen sobre la función renal. El primer paso es la unión de la 1,25 (OH)2-D3 a un receptor específico del citosol. Una vez ligado a su receptor, bajo la acción de la temperatura emigra al núcleo donde aumenta la síntesis de ARN. La 1,25-(HO)2D3 estimula la actividad de la ARN polimerasa II, enzima responsable de la formación de ARNm. Los nuevos mensajes inducirían en el polisoma la traslación de proteínas que alterarían la función celular aumentando su capacidad para transportar calcio y fósforo. Entre las proteínas que se forman bajo la acción de la 1,25-(HO)-D3 la que ha merecido mayor atención es la proteína ligadora de calcio(CaBP), ausente en los animales con carencia de vitamina D, que aparece después de la administración de la misma, y que se correlaciona con las modificaciones en la absorción intestinal de calcio. La CaBP no es la única proteína que se sintetiza en el intestino en respuesta a la 1,25-(HO)2 D3, se han identificado una fosfatasa alcalina que podría favorecer la traslación de fosfato y otras sintetizadas en las membranas del ribete en cepillo que aparecen más precozmente que la CaBP. FIGURA N°3 MECANISMO INTRACELULAR DE ACCION DEL 1,25 DIHIDROXICOLECALCIFEROL 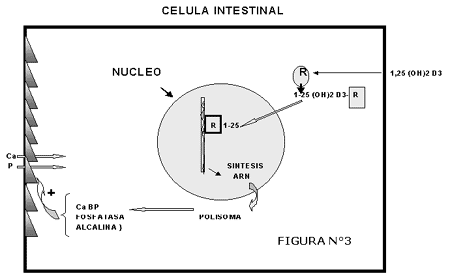 El primer paso es la unión de la 1,25-(OH)2-D3 a un receptor específico en el citosol. Una vez ligado a su receptor, la 1,25-(OH)2-D3 emigra al núcleo donde aumenta la síntesis nuclear de ARN. Esta vitamina eleva la actividad de la ARN polimerasa II, enzima responsable de la formación de ARNm. Los nuevos mensajes inducirán en el polisoma la traslación de proteínas que alterarían la función celular aumentando su capacidad para transportar Calcio y Fosfato. Entre las proteínas que se forman bajo la acción de la 1-25(OH)2-D3 la que merece mayor atención es la proteína ligadora de Calcio (Ca BP), encontrándo-se otra importante como la Fosfatasa alcalina, esta favorece la traslocación de fosfato en la membrana del ribete en cepillo y es sintetizada mas precozmente que la Ca BP. D - EXCRECIÓN DE CALCIO Y FÓSFORO Excreción Fecal El Ca fecal, fundamentalmente de origen dietario, puede estar dividido como exógeno o endógeno. El Ca exógeno es el Ca no digerido del alimento. El Ca endógeno es la fracción de Ca derivada de la secreción de Ca hacia el intestino, fundamentalmente hacia yeyuno. El conocimiento del Ca y PO4 fecal endógeno es importante para la determinación de la digestibilidad verdadera del Ca y PO4 del alimento. La digestibilidad aparente (Ca fecal en alimento/Ca en alimento) tiene una base válida para estimar la digestibilidad verdadera solo cuando el Ca y PO4 endógeno tiene muy poca importancia y constituye una pequeña parte de Ca y PO4 fecal. En la vaca el Ca fecal ha sido estimado en 4-7 gr./día, es directamente proporcional al Peso corporal y poco influenciado por cambios dietéticos de corta duración. Este valor es mayor en los animales adultos. El fósforo fecal endógeno fue estimado en vacas lactantes con valores de 10-14 gr./día, o sea, dos veces la cantidad del Ca endógeno. Excreción urinaria La excreción es regulada por PTH y Vitamina D. Parte del Ca y PO4 filtrado es reabsorbido en los túbulos renales. El fósforo se reabsorbe en la porción final del túbulo contorneado proximal debido a una actividad enzimática. El Ca tiene umbral de excreción y este oscila entre 6 a 8 mg%. Las dietas ácidas aumentan la excreción urinaria de Ca. En la vaca el rango de excreción urinaria de fósforo es solo una fracción del 1% del rango de excreción de fósforo en heces; esto es debido a la orina alcalina de los bovinos, pues con pH alcalino se encuentra limitada la secreción simultánea de calcio y fósforo. Secreción mamaria En vacas lactando, grandes cantidades de Ca y PO4 son secretadas en la leche, por ello muchas veces estos minerales son hallados en concentración más alta que en el plasma, el Ca 12 veces, el PO4 7 veces y el Mg 6 veces. FIGURA N°4 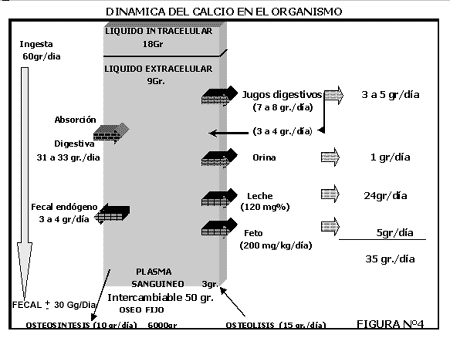 II- NIVELES DE CALCIO Y FÓSFORO SÉRICO Casi la totalidad del Ca que contiene la sangre se encuentra en el plasma; el PO4 y el Mg están localizados tanto en plasma como en los eritrocitos. El Ca y el Mg se encuentran: A- Ionizados: forma fisiológicamente activa, siendo más o menos el 40 a 60 % del total. B- No ionizados: en estado de unión con proteínas o como sales poco ionizables de ácidos como cítrico y fosfórico. La relación entre estas dos formas responde a la Ley de Acción de las Masas:  La proporción de Ca y Mg ligado a las proteínas depende de la concentración de las mismas, en particular de la albúmina, pues esta presenta una elevada capacidad de fijación para ambos iones. También influye la concentración de ambos iones y el pH del plasma. Si disminuye el pH la proporción de las formas iónicas aumenta.Además, está comprobado que el Ca unido a proteínas (BP-Ca) está formado por dos pooles que se distinguen por diferentes capacidades de unión con el Ca. El pool de reducida afinidad, interviene en el intercambio con el Ca++ iónico, por lo tanto, participa en el mantenimiento del nivel de Ca ionizado. El pool de elevada afinidad (TBC, Calcio Fuertemente Unido, o ACP, Complejo Albúmina-Calcio-Palmitato) no es intercambiable y no participa en el nivel de Ca++. Este último grupo tiene un importante rol en el transporte de Ca a través de la membrana, y en la captación de Ca por las células ósea, en el hueso hay una captación preferencial del Ca cuando es presentado como TBC ante el Ca iónico). FIGURA N° 5 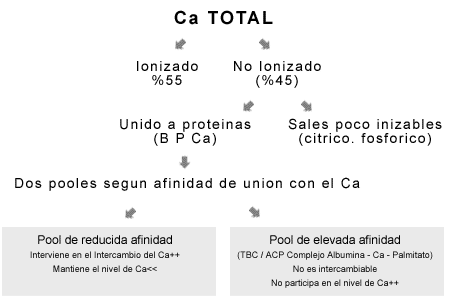 El Fósforo se encuentra en el plasma en tres formas: • Iones inorgánicos: PO4H21- y PO4H3-. • Unido a lípidos (30%). • Formando ésteres fosfóricos orgánicos. Las relaciones entre estas tres formas son consecuencia de otros factores aparte de simples equilibrios (como Ca y PO4). El pH tiene una gran influencia sobre la proporción en que se encuentran las dos formas de fosfato inorgánico en el plasma. Dicha relación es: 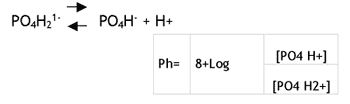 6.8 es el valor de pK para la disociación del PO4H21- bajo las condiciones que imperan en el suero. Al pH normal del plasma: [PO4H2-] = 4 x [PO4H2-] FIGURA N° 6 (DINAMICA DEL FÓSFORO EN EL ORGANISMO) 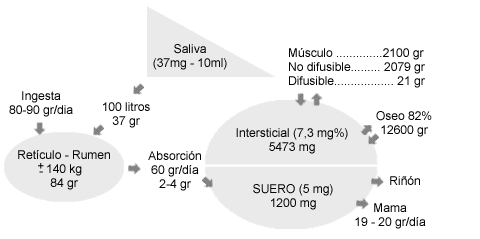 A - Hormona Paratiroidea La PTH es un polipéptido de 84 aminoácidos. Sin embargo, no es necesaria la hormona intacta para que ejerza su actividad biológica. Los fragmentos amino terminales 1-34 tienen la misma actividad que la hormona intacta, en cambio, los fragmentos con menos de 34 aminoácidos pierden esa actividad. Las células paratiroides sintetizan primero un precursor Pro-PTH con 6 aminoácidos adicionales amino terminales. La Pro-PTH es convertida en PTH en aproximadamente 24 minutos, tiempo que requiere su transporte por el Retículo Endoplasmático al Aparato de Golgi. Existe un depósito muy pequeño de PTH dentro de la célula. Apenas un 7% de este depósito de hallaría en forma de pro-PTH y normalmente no es liberada a la circulación; la pro-PTH carece de actividad biológica. Recientemente se ha descubierto que el mensajero ARN de la PTH modula la síntesis de un polipéptido mas largo que la pro-PTH, la pre-pro-PTH, del que 25 aminoácidos adicionales son hidrolizados pocos segundos después de su síntesis. Una vez sintetizada la PTH se acumula en los gránulos de secreción que atraviesan la célula, se unen a la membrana celular y liberan su contenido a la circulación. Este proceso de acumulación en gránulos secretorios hasta la liberación demora unos 30 minutos, pero posiblemente no sea imprescindible para la secreción de PTH, ya que la hormona podría ser secretada sin previa acumulación en gránu-los. (VER FIGURA N°7.) REGULACIÓN DE LA FUNCIÓN INTRACELULAR POR EL CALCIO El Calcio en sí tiene un pequeño efecto sobre el rango de síntesis de la Pro-PTH misma. El Calcio iónico regula el rango de degradación del péptido hormonal nuevo (Pro-PTH, PTH, o ambas) en la glándula durante la síntesis. La hipocalcemia es el principal estímulo para la secreción de PTH, el Ca a una concentración en suero, mayor a 2,5 mM (1,25 mM ionizado) inhibe la secreción de la hormona (2,5mM equivale a 10mg%). El Mg++ (Mg iónico) elevado inhibe y la disminución estimula la secreción de PTH. Esto ocurre en estado fisiológico normal, pero se observó que la administración de Mg ** produce en la glándula hiperactiva un aumento importante de la secreción. De todas maneras el ion Mg no ejerce un papel fisiológico en la regulación de la secreción de PTH. CONTROL DE LA SECRECIÓN. Las paratiroides deben aumentar rápidamente la concentración de PTH en res-puesta a la disminución de la concentración de calcio circulante, e inversamente, inhibir la liberación de hormona en respuesta en respuesta a un aumento indebido de la calcemia. La concentración extracelular de calcio iónico controla todos los pasos de síntesis y liberación hormonal. Uno de los más importantes sería la degradación intracelular de la hormona sintetizada. Una elevación del calcio iónico extracelular estimula y una disminución inhibe la degradación de PTH. Este mecanismo sería el responsable del rápido incremento de hormona liberada en respuesta a una hipocalcemia aguda, ya que la síntesis hormonal solo aumenta varias horas después del comienzo del estímulo. FIGURA N°7 (RUTAS DE BIOSÍNTESIS DE PTH Y CONTROL DE SECRECION DE PTH) 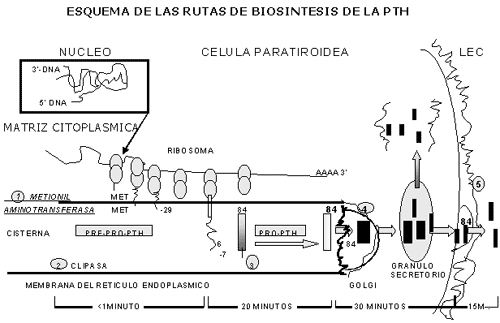 · La Pre-Pro-PTH es convertida en Pro-PTH por remoción del residuo metionil de NH2 terminal 1 por la Metil Aminotrasferasa, luego, la secuencia NH2 terminal (-29 a –7) de 23 aminoácidos es hidrolizada en segundos después de la síntesis 2. · Esta conversión ocurre durante el transporte del polipéptido dentro de la cisterna del Retículo Endotelial Rugoso. Veinte minutos después de la síntesis, la Pro-PTH llega a la región del Aparato de Golgi y es convertida en PTH por remoción del residuo NH2 terminal hexa-peptido, 3 La PTH es depositada en el gránulo secretorio y parcialmente degradada en la célula. 4 cuando la secreción es suprimida por un aumento en la concentración de Ca++, o 5 descargada en el LEC en respuesta a una disminución en la con-centración de Ca++. FIGURA N° 8 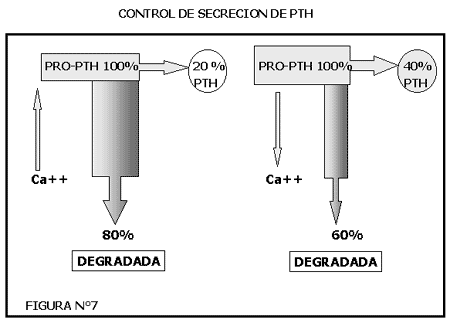 *La concentración de Ca++(iónico), controla tanto los pasos de síntesis como de liberación. Uno de los más importantes sería la degradación intracelular de la hormona sintetizada. Una disminución de Ca++ inhibe la degradación, mien-tras que una elevación la aumenta. Las variaciones en la secreción de PTH en respuesta a los cambios de la concen-tración de calcio no son lineales sino que representan una función sigmoidea de la calcemia. Dentro de lo que puede considerarse como los límites de concentración normal del calcio circulante (9 a 10,5 mg/100ml) la variación en la secreción de PTH es relativamente reducida. En cambio, la velocidad de secreción aumenta con rapidez al disminuir la calcemia por debajo de 9,0 mg/100ml. El ritmo máximo de secreción de PTH se logra al alcanzar la calcemia un nivel de alrededor de 6 mg/100ml, no obteniéndose mayor respuesta aunque la disminución de calcio continúe disminuyendo. FIGURA N°9 RELACION ENTRE LA CALCEMIA Y LA SECRECION DE PTH 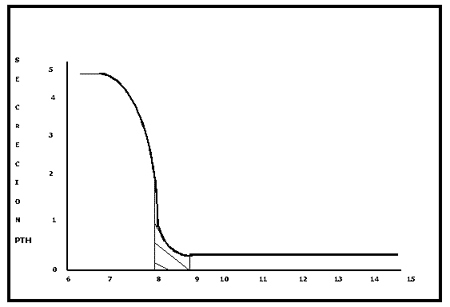 *El ritmo máximo de secreción de PTH se obtiene al alcanzar la calcemia un nivel de 6 mg/100 ml y no se obtiene mayor respuesta aunque la concentración de calcio continúe disminuyendo. En el otro extremo de la curva puede observarse que a partir de 11 mg/100 ml se obtiene la máxima inhibición en la secreción de PTH, sin embargo, esta inhibición máxima no alcanza nunca a suprimir por completo la secreción de hormona. La regulación ejercida por el calcio iónico sobre la secreción de PTH tiende a preservar, junto con otros mecanismos, los niveles de calcio extracelular. La curva de regulación descripta tiene una significación directa en las patologías de las glándulas paratiroides. En vacas con Hipoparatiroidismo nutricional la calcemia no desciende por debajo de los 5-6 mg/100 ml. Este nivel de calcio extracelular puede ser mantenido indefinidamente por el calcio liberado de la enorme reserva del organismo que es el esqueleto, sin necesidad de ningún estímulo hormonal. En síntesis, el estímulo para una mayor secreción de PTH inducida por la hipocalcemia alcanza un nivel máximo en un punto (alrededor de 6 mg/100ml) en el cual una mayor concentración hormonal no serviría para ningún fin específico. Por otra parte, la incapacidad del calcio iónico de suprimir por completo la secreción de PTH explica la fisiopatología del Hiperparatiroidismo con hipercalcemia. HORMONA PARATIROIDEA CIRCULANTE Y METABOLISMO PERIFÉRICO Desde a que se pudo medir PTH por radioinmunoanálisis se han identificado por lo menos tres fracciones diferentes de esta hormona, por lo tanto ahora sabe-mos que la misma no es homogénea en su composición química. La primera fracción o Pico I es una PTH de peso molecular de alrededor de 9.500 presumiblemente con la secuencia de aminoácidos 1-84. Esta fracción es biológicamente activa; su vida media en la circulación es muy corta, solo de unos pocos minutos, y contiene tanto el extremo NH2 como el COOH terminal. El pico II tiene un peso molecular de 7.500 aproximadamente, contiene solo el extremo COOH terminal, es biológicamente inactiva y su vida media en la circulación es de una hora. El pico III, con un peso molecular aproximado de 3.500, contiene principalmente el fragmento NH2 terminal biológicamente activo y con una vida media en la circulación de apenas 2 a 4 minutos. La presencia de los mencionados fragmentos de PTH en la circulación se debe al metabolismo periférico de la hormona que se efectúa en el hígado y en el riñón. La glándula secreta predominantemente la hormona intacta con la secuencia 1-84 de aminoácidos, aunque se admite que en determinadas circunstancia, principalmente en una hipercalcemia, puede liberar fragmentos a la circulación. La hormona intacta es metabolizada en el riñón, responsable del 60% de su destrucción, en el hígado también ocurre esta degradación. En este órgano se encuentran receptores que reconocen la zona 24-48 de aminoácidos intactos. Por lo tanto, el hígado extrae de la circulación solamente la hormona intacta, a partir de la cual forma el fragmento 1-34 o 1-37 NH2 terminal biológicamente activo y el fragmento 34-84 COOH terminal carente de actividad. Este proceso se produce en las células de Kupffer, ya que la vida media de la PTH intacta es sólo 12 minutos en estas células, mientras que es de 80 minutos en los hepatocitos. El hígado no extrae los fragmentos una vez que estos están en la circulación. Y lo más interesante es que la velocidad de conversión de la hormona intacta en sus fragmentos derivados depende de la concentración de Calcio en el medio de perfusión. Cuando la concentración disminuye a menos de 5 mg/100ml, la conversión se acelera, e inversamente por encima de 11 mg/100ml esa conversión se retarda. El riñón en cambio es capaz de metabolizar tanto la PTH intacta como sus fragmentos. Existen dos mecanismos distintos de degradación renal. 1. Por filtración glomerular y posterior reabsorción tubular y degradación intracelular, el riñón es capaz de metabolizar tanto la PTH intacta como sus fragmentos NH2 o COOH terminales. 2. Por extracción peritubular, las células peritubulares del riñón tienen receptores para la hormona para la hormona intacta y para el fragmento NH2 terminal - es necesaria la secuencia 25-34 -, los que serían metalizados y producirían los efectos renales de la hormona. No hay extracción renal peritubular de los fragmentos inactivos. El hueso, uno de los principales efectores de la hormona, no ejerce ninguna función en su metabolismo periférico. El tejido óseo se limita a extraer el fragmento NH2 terminal, que es el único con actividad biológica sobre aquel, pero no extrae de la circulación la hormona intacta, ni los fragmentos COOH terminales. En síntesis, la hormona intacta secretada por la glándula es hidrolizada en el hígado y en el riñón con formación de dos fragmentos, el NH2 terminal, con actividad biológica sobre todos los efectores y vida media muy breve, y el CO-OH terminal biológicamente inactivo, con vida media prolongada y solo degradable por filtración glomerular y reabsorción tubular en el riñón. La concentración de Calcio regularía la formación periférica de los fragmentos activos. La concentración de PTH biológicamente activa, es de alrededor de 11 pg/ml, con un rango de 3 a 29 pg/ml. FUNCION DE LA HORMONA PARATIROIDEA El papel fisiológico fundamental de la PTH es el de mantener un tenor adecuado de Calcio en el compartimento extracelular. Para cumplir esta función la PTH actúa. 1. sobre el esqueleto, a través de una serie de mecanismos complejos que efectúan el Modelamiento y Remodelamiento óseo y el pasaje de Calcio a través de la membrana ósea. 2. Sobre el riñón aumentando la resorción tubular de Calcio y disminuyendo la de Fósforo. 3. Sobre el metabolismo de la vitamina D, estimulando la síntesis del 1,25(OH)2 D3 y secundariamente, la absorción intestinal de Calcio. ACCION SOBRE EL HUESO Debemos repasar la fisiología ósea ( ya descripta) para comprender la acción de la hormona sobre el hueso. La PTH actuaría sobre tres procesos óseos: a) Sobre los osteocitos de superficie y el intercambio de Calcio entre el Líquido Extracelular Óseo (LEC) y el sistémico. b) Sobre los osteocitos de profundidad. c) Sobre el remodelamiento y Modelamiento óseos a través de los osteo-clastos y osteoblastos. La PTH aumenta el pasaje de Calcio del LEC óseo al sistémico. Esto podría producirse por el aumento en la producción de ácidos orgánicos que induce la PTH en los osteocitos, lo que a su vez aumenta la solubilidad del mineral y permite una mayor disponibilidad para su posterior liberación. Independientemente o asociado a este mecanismo la PTH aumenta la actividad de la bomba de Calcio, a través de la cual los osteocitos incrementan la salida de este mineral hacia el LEC sistémico. La PTH es, asimismo, por lo menos en dosis farmacológicas un activo vasodilatador arterial, de esta manera que aumenta el flujo arterial del hueso. Este efecto de la PTH podría ser necesario para asegurar un correcto flujo circulatorio del hueso en el momento en que se produce una mayor salida de Calcio a través de la membrana formada por los osteocitos de superficie. Alrededor de los osteocitos profundos existe un tejido óseo perilacunar poco estructurado y metabólicamente activo. Este metabolismo está regulado por los niveles de PTH. Un aumento rápido en la concentración de esta hormona determina una resorción de la matriz y disolución mineral. El incremento sostenido de la secreción de PTH lleva a aumentar el espacio perilacunar, con posibilidad de que estos espacios confluyan entre sí. Siempre se generalizó la idea de que las respuestas bioquímicas ocurren principalmente en los osteoclastos, en la actualidad se considera que no siempre es así, puesto que datos histológicos muestran que los osteocitos tienen aún una mayor participación en la osteólisis, sobre todo en las etapas tempranas. La respuesta mas temprana de las células óseas a la PTH es una entrada de los iones Ca++ y una activación de la adenilciclasa de la membrana. El Ca++ que entra actúa como segundo mensajero en la respuesta Osteolítica. (elevando la concentración de Ca++ en la célula se activa la osteólisis). Se aprecia incremento en la síntesis de RNA en el hueso, salida de enzimas lisosómicas y aumento de la actividad de esas enzimas en solución. También hay evidencias que la PTH estimula la formación de 3’-5’ AMPc en las células óseas y que éste es también un segundo mensajero en la respuesta Osteolítica. Este mensajero proviene de la activación de la adenilciclasa de la membrana. Se comprobó que el Ca++ y el AMPc desempeñan papeles independientes en la respuesta celular a la PTH. La concentración aumentada de Ca++ en el medio no estimula sino que inhibe la actividad de la adenilciclasa y el AMPc no induce al aumento de la permeabilidad al calcio, ya sea in vivo o en células aisladas. Un efecto conocido es la acción inhibitoria de la osteólisis ejercida por la calcitonina. Los últimos trabajos demuestran que la calcitonina no interviene en la entrada inicial de Ca++ o en la respuesta adenilciclásica de las membranas y tampoco en el bloqueo de la eliminación o activación de las enzimas Osteolítica. El mecanismo propuesto por Rasmussen es que la calcitonina puede disminuir la concentración de Ca++ activo no por interferencia en la entrada sino por estímulo de la toma del Ca++ mitocondrial Se ha comprobado que algunas quinasas - fosforiladas son totalmente independientes de la presencia del Ca++iónico. Algunas de las fosforilaciones activan enzimas almacenadas en formas precursoras; otras probablemente liberan y activan enzimas hidrolíticas de los lisosomas. Estos efectos bioquímicos acompañan a la osteólisis inducida por la PTH. Así se ve; síntesis acelerada de hialuronatos, Mucopolisacáridos sulfatados, acumulación de citratos y lactato, liberación de enzimas lisosómicas, activación de la anhidrasa carbónica y activación de fosfatasa ácida. Esta actividad se ha observado en los sitios de resorción activa. La vitamina D (1-25 (OH)2 D3) que actúa en el control de la absorción intestinal es muy activa también para inducir osteólisis. ACCION ARMONICA DE LA PTH EN EL REMODELAMIENTO OSEO Como ya desarrollamos en Fisiología ósea los osteoblastos y osteoclastos actúan armónicamente en el proceso de remodelamiento del hueso, al punto que han sido en conjunto denominados Unidad Cortical de Remodelamiento. Esta unidad actúa de la siguiente manera; los osteoclastos producen en la zona cortical túneles longitudinales que avanzan en sentido centrífugo y frente a los cuales los osteoblastos que forman la capa ósea van cerrando dichos conductos actuando en sentido centrípeto. El equilibrio armónico entre la destrucción y el crecimiento está regulado por el intercambio del líquido extracelular del hueso y el líquido extracelular sistémico. El pasaje del Ca++ se produciría desde este último al óseo por un proceso activo y los osteocitos se encargarían del proceso inverso. La existencia de este proceso activo, energéticamente dependiente, asegura, aún en ausencia de PTH una concentración de Ca++ de 6 mg/100ml en la circulación. La PTH actúa sobre los osteocitos de superficie obrando entonces en la modulación del intercambio del Ca++entre los líquidos extracelulares. Provoca el au-mento del pasaje del Ca++ óseo a la circulación. Claro está que para que el Ca++ pueda realizar este pasaje debe generarse ácidos orgánicos en los osteocitos, inducido por la PTH. Baja concentración de PTH activa los osteoclastos que están en reposo. Si dicha concentración aumenta hay mayor diferenciación de las células progenitoras a osteoclastos. Pero al mismo tiempo la PTH actúa sobre el proceso de la formación ósea. A dosis mayores disminuye el número de osteoblastos y por lo tanto la síntesis de colágeno, pero luego de unos días aumenta el número de osteoblastos y aquella síntesis. La consecuencia de todos estos procesos es que los osteocitos de superficie aumentan su transferencia de Ca++ desde el líquido extracelular del hueso hasta la circulación, que los osteocitos profundos provocan la resorción del tejido óseo perilacunar y que las células progenitoras se transforman en osteoclastos provocando el aumento de unidades de remodelamiento y el incremento de la velocidad de recambio del hueso. Por eso se produce, en última instancia, aumento de la resorción en la superficie endostal y estímulo de la formación a nivel perióstico. B) Tirocalcitonina La calcitonina o Tirocalcitonina es un Polipéptido compuesto por 32 Aminoácidos con un puente disulfuro en la posición 1-7, con un peso molecular (PM) de 3.600. Se produce en las células parafoliculares del tiroides, en las cuales se sintetiza la Pre-Pro-CalciTonina un Polipétido de 141 AA, que da lugar a la Pro-CT de 11 AA. Finalmente la Pro-CT. es procesada a Calcitonina de 32 AA y Catacalcina de 21 AA El metabolismo se lleva a cabo en el hígado y riñón. La pérdida de un solo AA produce la anulación de gran parte de su actividad biológica. Regulación de la secreción El principal estímulo es el aumento del tenor de Calcio en el Líquido extracelular (LEC). El Magnesio tiene un efecto similar, aunque en condiciones fisiológicas tiene poca acción. La Gastrina, Pentagastrina, Secretina, Glucagón, estimulan la secreción de TC. El alcohol es un potente estímulo, la respuesta es rápida y se produce tanto al ser administrado por vía endovenosa como oral. Los estrógenos estimulan la secreción de TC durante la preñez y alcanza un nivel máximo en el último tercio y en el post parto inmediato. Después de la ingestión de pastos ricos en Calcio, se produce una elevación momentánea de este mineral en plasma. La CT secretada en respuesta a la Hipercalcemia Post Prandial “facilita” la transferencia neta de Calcio al riñón y suprime transitoriamente la resorción del hueso. Esta acción sería necesaria en los estados del crecimiento (desarrollo post natal, crianza). Como la absorción de Calcio intestinal es la única ruta neta de entrada de Calcio al organismo y es durante el crecimiento cuando se necesita acumular gran parte del calcio que entra por esa vía, para soportar el crecimiento del esqueleto la, la CT juega en este punto un rol fundamental. Es sabido que la CT posee valores altos en la circulación fetal y que su secreción es más efectiva en los jóvenes para disminuir en el envejecimiento. Efectos de la calcitonina (ct) sobre el hueso La CT inhibe la resorción, acción que lleva a cabo reduciendo el número de Osteoclastos. Ellos se retraen y se separan de la superficie ósea, disminuyendo el número de sus núcleos. En los Osteoclastos existen millones de receptores a la CT en una magnitud 2 a 3 veces superior a la existente en otro tipo de células. En las vacas adultas donde la tasa de recambio es moderada la CT no produce una marcada hipocalcemia. La CT provoca una disminución en el tamaño de los Osteoclastos de profundidad, los núcleos se hacen picnóticos, de esta manera disminuye la actividad Osteocítica Periostiocitaria. La CT estimula el desarrollo del cartílago de crecimiento, aumenta la producción de glucoproteína y de Colágeno óseo en los Osteoblastos. Efectos de la calcitonina sobre el riñón La hormona aumenta la eliminación de calcio y Fósforo contribuyendo a la Hipocalcemia e Hipofosfatemia. Por lo tanto su acción es semejante a la PHT con respecto al Fósforo y antagónica con el Calcio. Aumenta la excreción renal de Na+, K+ y Mg++. La CT aumenta la captación de Fosfatos por los tejidos blandos, especialmente hígado. Integración de la regulación hormonal en el control del metabolismo del calcio y fósforo Una disminución en la Calcemia provoca un aumento de la liberación y síntesis de PTH. Esta actúa en forma medianamente rápida sobre la resorción tubular, disminuyendo la excreción de Calcio y aumentando la de Fosfatos. La PHT actúa también sobre el hueso aumentando el pasaje de Calcio a través de la membrana que separa el LEC óseo del sistémico, incrementando la Osteólisis alrededor de los Osteocitos. Esta respuesta “combinada” sería suficiente para que la Calcemia retornara a su nivel normal, ante estímulos “poco intensos y prolongados”. En caso contrario continúa la secreción de PHT y se ponen en juego mecanismos que prolongan y acentúan la acción hormonal. Sobre el hueso se produce un aumento de la actividad del número de Ostoclastos con activación de nuevas Unidades Corticales de Remodelamiento Óseo (UCR), a su vez comienzan a aparecer los efectos de la 1-25(OH)2 D3, aumentando la resorción intestinal de Calcio, reforzando también la activación de la PHT sobre la resorción Ósea. Es de primordial importancia para el mantenimiento de la acción de la PHT la existencia de un nivel adecuado de “mineralización ósea”, de allí que si no existe actividad de Vitamina D no hay acción de PHT. Fundamentales son entonces los efectos combinados de 1-25(OH)2D3 y PHT. La Figura N°9 nos demuestra la integración del mecanismo hormonal, allí podemos ver como la regulación de la Calcemia es un mecanismo estrechamente ligado al equilibrio de dichas hormonas. Siendo el Calcio un mineral estrechamente regulado, se puede predecir una futuro desequilibrio cuando los valores de este mineral varían ampliamente de un animal a otro. Las figuras 10 y 11 muestran la integración de la Vitamina D y sus metabolitos con la PTH y Tirocalcitonina i en la regulación de la Calcemia. FIGURA N° 10 Consecuencias de una Hipocalcemia 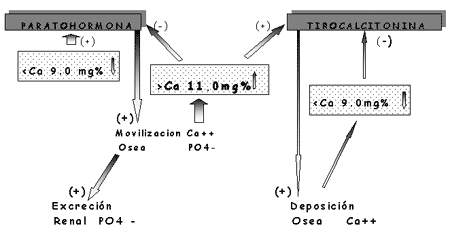 FIGURA N°11 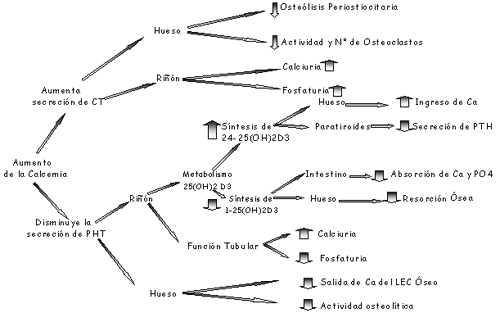 FIGURA N°11 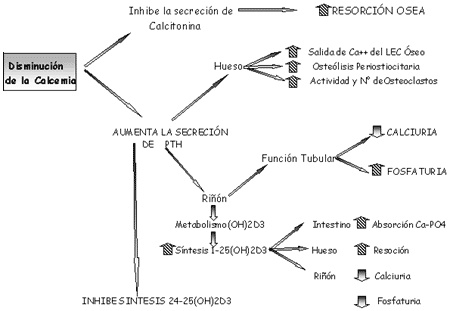 |
Anticuerpo monoclonal para RANKL.
13 de abril de 2010 - Tokio, Japón - Oriental Yeast Company, matriz de OYC Americas, se complace en anunciar que su anticuerpo monoclonal RANKL antimurino fue usado en el estudio "Control of mammary stem cell function by hormone signaling" (Control de la función de la célula madre mamaria mediante señalización hormonal) que fue publicado por Nature, una publicación internacional semanal de ciencias. El anticuerpo monoclonal RANKL antimurino fue desarrollado por el Instituto Nagahama para ciencias bioquímicas de OYC, Nagahama, Japón. El ligando del receptor activador NF-kB (RANKL), también llamado TRANCE, OPGL y ODF, es miembro de la familia TNF que se expresa en osteoblastos, células T activadas. RANKL interactúa con su receptor RANK expresado en progenitores de osteoclastos y células dendríticas maduras, llevando a la osteoclastogénesis y a la proliferación de células T, respectivamente. Resumen El mejor conocimiento de la fisiopatología ósea ha llevado a encontrar nuevas dianas terapéuticas en osteoporosis y se ha conseguido bloquear alguna de ellas con anticuerpos monoclonales. El sistema RANK-RANKL-OPG se considera el elemento efector final de los factores reguladores de la resorción ósea. El denosumab es un anticuerpo monoclonal completamente humanizado (IgG2) con una alta afinidad por el RANKL. Al unirse al RANKL mimetiza la acción de la OPG, impide la interacción RANK-RANKL, bloquea la activación de los osteoclastos e inhibe la resorción ósea. Denosumab ha demostrado en varios ensayos fase III que es un rápido, potente y seguro agente antirresortivo. Administrado por vía subcutánea cada 6 meses, aumenta la densidad mineral ósea y se acompaña de una rápida reducción de los marcadores de resorción ósea. Según los resultados del estudio FREEDOM en mujeres posmenopáusicas con osteoporosis, el tratamiento con 60mg/sc de denosumab cada 6 meses durante 3 años se acompaña de una reducción de riesgo relativo de fractura vertebral del 68% (incidencia del 2,3% en pacientes tratadas con denosumab y del 7,2% en las que recibieron placebo), de fracturas no vertebrales del 20% (incidencia de 6,5% con denosumab frente al 8% con placebo) y de fracturas de cadera del 40% (incidencia de 0,7% con denosumab frente a un 1,2% con placebo). Es un fármaco seguro, con una frecuencia de efectos adversos similares al placebo, aunque parece existir un aumento de efectos adversos cutáneos. Se está investigando el bloqueo mediante anticuerpos monoclonales de antagonistas de la vía de señalización Wnt/β-catenina, y en concreto la utilidad de los anticuerpos antiesclerostina y de los anticuerpos anti-Dkk. Este bloqueo de la vía Wnt/β-catenina tendría una acción anabólica sobre el remodelado óseo. Palabras clave Osteoporosis. Anticuerpos monoclonales. Denosumab. Texto completoObtener mejores tratamientos para la osteoporosis es uno de los actuales retos de la medicina. Los recientes descubrimientos en la fisiopatología ósea han llevado a encontrar nuevas dianas terapéuticas1 y en los últimos años se ha realizado un amplio esfuerzo para lograr bloquear alguna de estas dianas con anticuerpos monoclonales. Los frutos de este trabajo han llegado a buen fin y ya es posible bloquear el sistema RANK-RANKL-OPG con denosumab (AMG 162), un anticuerpo monoclonal con una potente acción antirresortiva2. Para un futuro no muy lejano se está investigando el bloqueo mediante anticuerpos de alguno de los antagonistas de la vía de señalización Wnt/β-catenina, y en concreto la utilidad de los anticuerpos antiesclerostina (AMG 785) y de los anticuerpos anti-Dkk13, 4. Este bloqueo de la vía Wnt/β-catenina tendría una acción anabólica sobre el remodelado óseo. Sistema RANK-RANKL-OPGEl sistema del RANK-RANKL-OPG tiene un papel crucial en la regulación de la resorción ósea y del remodelado óseo5, 6, 7. Desde hace varios años se ha intentado obtener agentes terapéuticos capaces de actuar sobre la vía del RANK-RANKL-OPG. El RANKL es una proteína transmembrana que se encuentra en la superficie de las células de la línea osteoblástica, así como en una forma soluble liberada por proteólisis. También se expresa en otras células, como linfocitos, células mamarias, células dendríticas, osteoclastos, sinoviocitos y en células cancerosas de la mama y de la próstata. En presencia del factor estimulante de colonias de macrófagos (M-CSF), el RANKL es un mediador fundamental en la formación, función y supervivencia de los osteoclastos8. El RANKL se une a su receptor RANK en los osteoclastos y provoca su maduración hacia osteoclastos activados. El RANK es un miembro de la familia de receptores del TNF-α y se expresa en los precursores de osteoclastos y en osteoclastos. La osteoprotegerina (OPG) es un inhibidor endógeno natural del RANKL y actúa como un receptor señuelo. La OPG se une al RANKL y neutraliza sus efectos, inhibiendo la resorción ósea9. La OPG actúa como un inhibidor de la función y diferenciación de los osteoclastos. El presente: bloqueo del RANKLDesde 1997 se han evaluado diferentes compuestos dirigidos a bloquear al RANKL. Inicialmente, se utilizaron proteínas de fusión en estudios preclínicos y clínicos. En las proteínas Fc-OPG y OPG-Fc, la OPG se fusionó con la porción Fc de la IgG1; la molécula RANK-Fc se formó a partir de la fusión del dominio extracelular del RANK con la porción Fc de la IgG1. Más tarde se investigó un anticuerpo monoclonal totalmente humano dirigido al RANKL, el AMG 162, obtenido a partir de ratones transgénicos10. Estos ratones se generan mediante la sustitución de genes Ig murinos por Ig humanos. Teóricamente, los anticuerpos obtenidos tienen una menor propensión a desarrollar respuestas anti-anticuerpo. En los estudios preclínicos realizados con ratones, y debido a su gran especificidad para el RANKL humano, denosumab no suprime la resorción ósea. En ratonesknockout que expresan RANKL quimérico, denosumab reduce la resorción ósea y aumenta la masa ósea11. Sólo se continuó con el desarrollo clínico del AMG 162 (denosumab) por sus mejores características. Aunque todas las moléculas eran eficaces, denosumab presentaba mejores propiedades, con una mayor actividad y duración del efecto antirresortivo. Además, tenía ventajas de seguridad; en los estudios no se detectaron anticuerpos neutralizantes. Por otro lado, denosumab no se une al TNF-α, al TNF-β, al ligando CD40 ni al ligando relacionado con el inductor de apoptosis del TNF (TRAIL)10. Denosumab: características farmacocinéticasDenosumab es el primer agente biológico aprobado para el tratamiento de la osteoporosis. Se une con gran afinidad y especificidad al RANKL humano, mimetiza la acción de la OPG y reduce la resorción ósea7. La unión de denosumab al RANKL se ha investigado in vitro mediante citometría de flujo y ELISA. Denosumab se une tanto a la forma soluble como a la forma transmembrana. Esta unión es inhibida por un exceso de RANKL, pero no de TNF-α, TNF-β, TRAIL o del ligando CD40. Denosumab tiene una farmacocinética no lineal, similar a otros anticuerpos monoclonales humanos. Tras su administración subcutánea (sc) se absorbe por vía linfática para llegar al torrente sanguíneo. Presenta una primera fase de absorción, con una Cmáx entre los días 5-21 según la dosis utilizada, una segunda fase con una semivida media de 32 días para las dosis altas y una fase de descenso rápido cuando la concentración está por debajo de 1.000 ng/ml. El aclaramiento se produce a través el sistema retículo endotelial y no tiene excreción renal. La administración sc de denosumab produce una reducción rápida, entre 12 y 72h, y dependiente de la dosis de la función osteoclástica, que se mantiene durante 6 meses como pone de manifiesto la supresión de los marcadores de resorción. En un estudio fase I en mujeres posmenopáusicas12, se estudió el efecto de una dosis única de denosumab o placebo sobre los marcadores de remodelado óseo (MRO). Se observó una reducción dependiente de la dosis del telopéptido N-terminal (NTX), un marcador de resorción ósea. Esta reducción fue rápida; a los 3 meses alcanzaba el 84% respecto al valor inicial, se mantuvo cerca de 6 meses y fue reversible tras la retirada del fármaco. La reducción en los marcadores de formación fue más tardía y menos pronunciada. Denosumab y densidad mineral óseaEn un estudio de fase II, doble ciego y controlado con placebo, un total de 412 mujeres posmenopáusicas con una densidad mineral ósea (DMO) baja fueron aleatorizadas a recibir varias dosis de denosumab cada 3 o 6 meses, alendronato semanal o placebo. La variable principal de desenlace fue el cambio en la DMO lumbar a los 12 meses. En el grupo tratado con denosumab se observó un incremento significativo de la DMO lumbar, entre un 3,0 y un 6,7% según dosis y pauta de dosificación. El aumento en el fémur total fue del 1,9 al 3,6% y en el radio distal del 0,4 al 1,3%. El tratamiento con denosumab produjo una reducción rápida, dependiente de la dosis, de los MRO, que se mantuvo durante los 12 meses13. Resultados de estudios de extensión confirman estos mismos resultados, tanto para el aumento de la DMO como en la reducción de los MRO. La prolongación del estudio a 4 años y 6 años muestra un aumento progresivo de la DMO. A los 4 años el tratamiento con denosumab se asoció con un aumento entre el 9,4 al 11% en la columna lumbar y del 4 al 6,1% en el fémur total. El tratamiento durante 6 años con denosumab 60mg/sc cada 6 meses se acompañó de un aumento de la DMO del 13,3% en la columna lumbar14. El estudio DEFEND es un estudio internacional controlado con placebo de 2 años de duración realizado para conocer el efecto de denosumab sobre la DMO en mujeres con osteopenia. Participaron 332 mujeres posmenopáusicas con edades entre 65 y 90 años, y valores del T-scoreen columna lumbar entre –1 y –2,5. El objetivo del estudio fue evaluar el cambio en la DMO lumbar después de 2 años de tratamiento. Comparado con placebo, el tratamiento con denosumab 60mg sc cada 6 meses aumentó de forma significativa la DMO en la columna lumbar (6,5% vs –0,6%) y redujo los MRO15. En un subestudio que analizaba los cambios en la DMO mediante pQTC del radio, el tratamiento con denosumab se acompañó de un aumento de la DMO y de la mejoría en parámetros de resistencia ósea16. Eficacia antifractura del denosumabEl estudio FREEDOM es un ensayo clínico internacional en fase III, aleatorizado y controlado con placebo, de 3 años de duración, realizado para evaluar la eficacia de denosumab en la reducción de fracturas17. Se incluyeron a 7.868 mujeres posmenopáusicas con edades comprendidas entre los 60 y los 90 años. Todas las mujeres tenían una DMO en la columna lumbar y/o fémur total, con un T-score entre –2,5 y –4. Se excluyó a las pacientes con fractura vertebral grave, dos o más fracturas de grado moderado o concentraciones de 25(OH)D3 por debajo de 12 ng/ml. Las pacientes recibieron denosumab 60mg sc cada 6 meses o placebo. Aproximadamente un 24% tenía una fractura vertebral prevalente. El riesgo medio de fractura principal a 10 años según FRAX fue del 18,3% para el denosumab y del 18,4% para el placebo. Todas las pacientes recibieron 1g de calcio y dosis variables de vitamina D, según la concentración de 25(OH)D3. El objetivo principal era la reducción de nuevas fracturas morfométricas vertebrales a los 3 años. Comparado con placebo, denosumab redujo la incidencia a los 3 años de nuevas fracturas vertebrales del 7,2 al 2,3% (reducción del riesgo relativo del 68%). Para la fractura de cadera la reducción a 3 años fue del 1,2 al 0,7%, lo que representa una reducción del riesgo relativo del 40%. La reducción del riesgo relativo de fractura no vertebral fue del 20%, con una reducción de la incidencia a 3 años del 8,0% al 6,5%17. La reducción del riesgo de fractura se acompañó de un aumento de la DMO a los 36 meses del 9,2% en columna lumbar y del 6% en fémur total, y de una reducción de los MRO. Estudios comparativos de denosumab con alendronatoSe han realizado varios estudios de no inferioridad para comparar el efecto de denosumab frente a alendronato sobre la DMO. No hay estudios comparativos sobre la reducción de fracturas. El estudio DECIDE es un estudio en fase III, aleatorizado y doble ciego, para determinar si existen diferencias en la DMO de fémur total en mujeres posmenopáusicas con baja DMO que reciben denosumab respecto de las que reciben alendronato semanal. Se incluyó a 1.189 mujeres, con una edad media de 64 años y un T-score ≤2,0 en la columna lumbar o en el fémur total18. Las pacientes se aleatorizaron para recibir denosumab 60mg sc cada 6 meses más placebo oral o alendronato 70mg semanal más inyecciones sc de placebo cada 6 meses. La variable principal fue el cambio en la DMO en fémur total a los 12 meses. Otras variables analizadas fueron los cambios en la DMO de columna, el cuello femoral, el trocánter y el tercio distal del radio y los cambios en los MRO. A los 12 meses, el tratamiento con denosumab comparado con alendronato dio lugar a aumentos significativamente mayores de la DMO en el fémur total (3,5% vs 2,6%), la columna lumbar, el trocánter, el cuello femoral y el tercio distal del radio. El estudio STAND es un estudio fase III internacional, aleatorizado, doble ciego, de grupos paralelos y de 1 año de seguimiento, con el objetivo de comparar la eficacia y seguridad de denosumab en pacientes tratados previamente con alendronato19. Se incluyó a 504 mujeres posmenopáusicas previamente tratadas con alendronato y se evaluaron los efectos del cambio a denosumab sobre la DMO y los MRO respecto a seguir el tratamiento con alendronato. La variable principal fue el cambio de la DMO en el fémur total a los 12 meses. Un objetivo secundario fue la seguridad de denosumab respecto al alendronato. Las pacientes tenían una masa ósea baja, con un T-score ≤ –2,0 y ≥ –4,0 en la columna o el fémur total. Las pacientes que pasaron a ser tratadas con denosumab tuvieron un mayor incremento de la DMO en la columna, el fémur total y el radio distal. La DMO en el fémur total aumentó en un 1,90% en las pacientes tratadas con denosumab frente al 1,05% en las que continuaron con alendronato. El perfil de seguridad entre ambos grupos de tratamiento fue similar. Seguridad del denosumabLos resultados publicados de los estudios en fase I y fase II muestran una frecuencia similar de efectos adversos en los grupos placebo, denosumab y alendronato, que fue el fármaco comparador activo12, 13, 14. En los estudios en fase III se aprecia un buen perfil de seguridad, sin diferencias significativas entre placebo y denosumab. Así en el estudio FREEDOM se observa una incidencia similar de infecciones oportunistas, tumores malignos y fallecimientos entre los grupos denosumab y placebo17. No se detectó en ningún caso el desarrollo de anticuerpos frente a denosumab. En cuanto a la aparición de infecciones, no se observa un aumento en el número total de infecciones, en infecciones de las vías respiratorias o en infecciones graves en los pacientes tratados con denosumab. El tratamiento con denosumab parece aumentar la incidencia de infecciones de la vía urinaria baja. Existe una mayor frecuencia de dolor de garganta y lesiones cutáneas, como eczema, erisipela y celulitis en los pacientes que recibieron denosumab. Estos efectos adversos podrían estar relacionados con un efecto directo de denosumab sobre el sistema inmunitario. El riesgo de hipocalcemia con denosumab es mayor en los pacientes que están en diálisis o que tienen un aclaramiento < 30ml/min. El tratamiento con denosumab está contraindicado en los pacientes con hipocalcemia. Desde el punto de vista práctico, debe prestarse especial atención en asegurar una ingesta adecuada de calcio y vitamina D. Los efectos adversos más frecuentes durante el tratamiento con denosumab (1-10%) recogidos en la ficha técnica fueron infecciones del tracto urinario, infecciones de las vías respiratorias, dolor ciático, cataratas, estreñimiento, erupción cutánea y dolor en brazos o piernas. Los casos de cataratas han aparecido sobre todo entre varones que reciben denosumab por cáncer de próstata. Se ha descrito algún caso aislado de osteonecrosis de mandíbula20. De manera similar a lo que ocurre con los bisfosfonatos, la utilización de denosumab a dosis elevadas en enfermos oncológicos se acompaña de la aparición de casos de osteonecrosis de mandíbula, con una incidencia cercana al 2%. La mayoría de los casos aparecen al utilizar dosis mensuales de 120mg21. No se han comunicado casos de fractura atípica del fémur ni de retraso en la consolidación de fracturas en pacientes con osteoporosis tratados con denosumab. Denosumab y artritis reumatoideDado el papel que tienen los osteoclastos en la erosión articular, el tratamiento con denosumab podría ser útil para prevenir el daño estructural en la artritis reumatoide. La destrucción ósea parece estar relacionada con los factores que inducen la activación de los osteoclastos, como IL-6, IL-11, IL-17 y, especialmente IL-1, TNF y RANKL. En modelos animales de artritis el bloqueo de RANKL detiene tanto la progresión del daño radiológico, como la pérdida ósea yuxtaarticular. En estos modelos la administración de OPG no inhibe la inflamación sinovial ni la neoformación ósea asociada a la inflamación. Cohen et al22 han publicado un estudio en fase II realizado para evaluar el efecto de denosumab sobre el daño estructural en pacientes con artritis reumatoide. Se incluye a 227 pacientes tratados con metotrexato y se establecen tres grupos de tratamiento: denosumab 60 sc cada 6 meses, denosumab 180mg sc cada 6 meses y un grupo control. Para estudiar el daño estructural se realiza una RM de la mano al inicio y después de 6 meses de tratamiento; los cambios en las erosiones por RM se miden mediante la puntuación RAMRIS propuesta por OMERACT. En pacientes con artritis reumatoide y tratados con metotrexato, denosumab parece detener la progresión del daño estructural y se observan menos erosiones articulares por RM. El tratamiento con denosumab reduce los MRO, detiene la pérdida ósea yuxtaarticular y aumenta la DMO de la mano medida por DXA23. Denosumab y cáncerUna posible indicación del tratamiento con denosumab es la osteopenia asociada a tratamientos hormonales en pacientes con cáncer. El estudio HALT analiza el efecto de denosumab sobre la DMO en 1.468 varones en tratamiento antiandrógenico por cáncer de próstata y alto riesgo de fractura24. Se trata de un estudio doble ciego y controlado con placebo. El objetivo principal fue el cambio en la DMO lumbar a los 3 años. Se observa un aumento significativo de DMO lumbar y femoral en el grupo tratado con denosumab. A los 36 meses, los pacientes del grupo de denosumab tenían una incidencia de nuevas fracturas vertebrales del 1,5 frente al 3,9% en el grupo placebo, lo que supone una reducción del riesgo relativo del riesgo de fractura vertebral del 62% a 3 años24. Recientemente se han publicado los resultados de dos estudios comparando el efecto de denosumab frente a zoledronato en la progresión de la metástasis en pacientes con cáncer de próstata resistente a la castración25 y en pacientes con cáncer de mama avanzado21. La variable principal fue la aparición de algún evento óseo relacionado, ya fuera una fractura patológica, la compresión medular o la necesidad de radioterapia o cirugía por metástasis ósea. En los dos estudios el tratamiento con denosumab retrasó la aparición de los eventos óseos relacionados con la metástasis. Hacia el futuro. Activación de la vía Wnt/β-cateninaAnticuerpos antiesclerostinaLa esclerostina es una proteína codificada por el gen SOST capaz de inhibir la osteoblastogénesis26. Mutaciones de la proteína LRP5 impiden la interacción normal entre la esclerostina y el correceptor de Wnt, y provocan un síndrome de masa ósea alta. La falta de expresión de esclerostina por mutaciones del gen SOST es la causa de la enfermedad de Van Buchem y de la esclerosteosis; estas displasias esqueléticas se caracterizan por un importante aumento de la DMO. Estos hallazgos en diferentes trastornos genéticos ha llevado a estudiar el bloqueo o la inactivación de la esclerostina como una posible diana terapéutica anabólica, a través de la vía Wnt, en la osteoporosis3. El tratamiento con anticuerpos monoclonales humanizados frente a la esclerostina (AMG 785)estimula la vía de señalización Wnt/β-catenina y aumenta la masa ósea en estudios en ratones y en primates. En ratas ovariectomizadas, la administración de anticuerpos antiesclerostina revierte la pérdida ósea, aumenta la masa ósea trabecular y mejora las propiedades estructurales del hueso3. Recientemente, se ha publicado un estudio clínico fase I en humanos controlado con placebo. La administración de una dosis única de anticuerpos antiesclerostina (AMG 785) en sujetos sanos, comparado con el placebo, se acompaña de un aumento de la DMO, un aumento de los marcadores de formación ósea y la reducción en los marcadores de resorción ósea27. Los anticuerpos antiesclerostina tienen un efecto anabólico cuando se administran a mujeres posmenopáusicas. Parece ser un tratamiento seguro y bien tolerado. En el estudio publicado se recoge un caso de hepatitis tóxica y la aparición en varios casos de anticuerpos neutralizantes. Se están realizando estudios de fase II para evaluar el efecto del anticuerpo antiesclerostina (AMG 785) sobre la DMO en mujeres posmenopáusicas con baja densidad ósea y también sobre la consolidación de fracturas. Anticuerpos anti-Dkk1Los antagonistas de Wnt, como las proteínas Dickkopf (Dkk), tienen un papel determinante en la regulación del remodelado óseo y en la pérdida de hueso asociada a la inflamación. Anticuerpos capaces de bloquear a Dkk1 pueden tener un papel protector frente a la pérdida ósea y las erosiones4. Se ha estudiado en un modelo animal de artritis si la estimulación de la vía Wnt con un anticuerpo anti-Dkk1 altera la pérdida ósea mediada por la inflamación articular. En ratones transgénicos para el TNF humano —un modelo animal de artritis erosiva— el tratamiento con anticuerpos anti-Dkk1 inhibe la pérdida ósea inducida por el TNF, sin alterar la inflamación sinovial28. La modulación de la vía Wnt/β-catenina mediante el bloqueo de Dkk1 puede ser un tratamiento útil frente a la enfermedad ósea asociada al mieloma y actualmente se está realizando un estudio fase I en pacientes con mieloma múltiple. |
fin hasta tercera parte.
Patologias varias... entrega de cuarta parte:
Raquitismo y osteomalacia
¿Qué es la osteomalacia y el raquitismo?en directorio de la a a la z de entorno medico
Ambos son defectos de la mineralización ósea. La primera (osteomalacia=huesos suaves) es la forma de raquitismo del adulto y por tanto el raquitismo como tal es de la infancia.
CAUSAS Y EPIDEMIOLOGÍA
La principal característica del raquitismo y de la osteomalacia es la falta de calcio en los huesos; el raquitismo ataca a los niños cuyos huesos todavía están en crecimiento, y la osteomalacia a los adultos que tienen los huesos formados. Ambos trastornos son producidos principalmente por una carencia de vitamina D y no por una falta dietética de calcio. Según se vio en los Capítulos 10 y 11, la vitamina D se obtiene de alimentos animales en la dieta y de la exposición de la piel a la luz solar. La vitamina D funciona como una hormona para regular el metabolismo del calcio.
Debido a que el cuerpo puede obtener cantidades adecuadas de vitamina D, inclusive a partir de una exposición moderada a la luz solar, el raquitismo y la osteomalacia son poco comunes en la mayoría de los países africanos, asiáticos y latinoamericanos, pues allí la luz solar es abundante. Cuando existen, se deben, por lo general, en parte a una práctica cultural particular o a ciertas circunstancias locales. Por ejemplo, en algunas sociedades musulmanas, las mujeres que practican el purdah usan ropas que cubren la mayor parte de la piel, y raramente salen del hogar con sus bebés. Se tiene información que el raquitismo en algunas ciudades grandes densamente pobladas (por ejemplo, Calcuta, India; Johannesburgo, Sudáfrica; Addis Abeba, Etiopía), ataca a los niños que no salen a la luz solar. Sin embargo, en ninguna parte de los trópicos o subtrópicos el raquitismo es una enfermedad de prevalencia alta, como lo fue en Europa en el siglo XIX (véase el Capítulo 11). En la actualidad, en el Reino Unido, el raquitismo y la osteomalacia se diagnostican en familias inmigrantes de origen asiático.
Casi siempre el raquitismo grave se presenta en niños menores de cuatro años de edad, que consumen pocas cantidades de alimentos de origen animal y que por algún motivo no tienen mucha exposición a la luz solar. Las deformidades óseas, sin embargo, pueden ser más obvias en niños mayores. La osteomalacia es más común en mujeres con varios niños, y que, como resultado de embarazos sucesivos y lactancia, llegan a tener agotamiento de calcio y e insuficiente vitamina D.
MANIFESTACIONES CLÍNICAS
Raquitismo
Raquitismo
Los niños con raquitismo, a diferencia de los que sufren tantas otras enfermedades carenciales, por lo general son rollizos y aparentan estar bien alimentados, debido a que su consumo de energía es casi siempre correcto. Su aspecto engaña a menudo a la madre quien piensa que todo está bien. Sin embargo, el niño se puede sentir indispuesto, y un examen más cuidadoso revelará la poca tonicidad muscular, que causa un abdomen protuberante. Otra característica del raquitismo es una alteración general del desarrollo normal. El niño se demora en alcanzar las etapas de la primera infancia, como la dentición aprender a sentarse y caminar. Otros síntomas generales incluyen molestias gastrointestinales y excesivo sudor en la cabeza.
Los signos principales de la enfermedad, y en los que se basa el diagnóstico de raquitismo son las deformaciones óseas (Foto 33). La primera y principal característica, es la hinchazón de los extremos en crecimiento (epífisis), de los huesos largos. Esta inflamación primero se puede encontrar en la muñeca, donde se afecta el radio. Otro sitio clásico es la unión de las costillas con el cartílago costal; aquí la inflamación tiene la apariencia de un rosario que se conoce como «rosario raquítico». También se pueden observar hinchazones en los pies, la tibia, el peroné y el fémur. En los bebés con raquitismo la fontanela anterior se cierra tardíamente y en los niños mayores no es rara una protuberancia del frontal.
Una vez que un niño con raquitismo empieza a pararse, a caminar y estar activo, desarrolla nuevas deformidades debido a la característica blanda y débil de los huesos. La deformidad más común son las piernas en arco (Foto 34); con menos frecuencia se ven las rodillas juntas. Más serias, sin embargo, son las deformidades de la columna vertebral. Los cambios en la pelvis, aunque raramente son visibles, pueden ocasionar dificultades en el parto a las mujeres que han sufrido raquitismo en la infancia.
El raquitismo se puede diagnosticar por la apariencia clínica y radiológica de los huesos, y por exámenes de laboratorio.
Osteomalacia
La osteomalacia se caracteriza por dolor, algunas veces grave, sobre todo en los huesos de la pelvis, la parte baja de la espalda y en las piernas. En ciertas ocasiones puede haber sensibilidad dolorosa en las tibias y otros huesos. El paciente casi siempre camina con los pies muy separados y parece caminar como pato. Las deformidades de la pelvis pueden ser evidentes. No son raros los espasmos tetánicos que se manifiestan por contractura involuntaria de los músculos de la cara o por espasmo carpopedal (donde hay en la mano un espasmo rígido con el pulgar que presiona la palma). Las fracturas espontáneas pueden ser una característica. Antes que las deformidades sean clínicamente demostrables, el diagnóstico se puede hacer por radiografías que mostrarán un enrarecimiento o descalificación de los huesos en todo el cuerpo. La osteomalacia no se debe confundir con la osteoporosis, enfermedad del envejecimiento, en la que la descalcificación también es una característica.
HALLAZGOS DE LABORATORIO
Los niveles en la sangre tanto de esteroles como de metabolitos de la vitamina D, que ahora se pueden medir en laboratorios especializados, son siempre muy bajos en casos de raquitismo y osteomalacia. También se observan niveles bajos de fósforo sérico y niveles altos de fosfatasa alcalina. Por lo general la cantidad de calcio en la orina es baja.
TRATAMIENTO
Raquitismo
Raquitismo
La base del tratamiento es suministrar vitamina D y calcio. La vitamina D puede administrarse como aceite de hígado de bacalao. Son suficientes tres cucharaditas tres veces al día que suministran aproximadamente 3 000 UI. También se puede utilizar calciferol sintético. El calcio es mejor darlo como leche, por lo menos medio litro al día. La leche de vaca contiene 120 mg de calcio por 100 ml.
Existe disponibilidad de tabletas que contienen vitamina D y calcio. A un niño menor de cinco años se le puede suministrar una tableta dos veces al día, y a uno mayor de esa edad una tableta tres veces al día.
Mientras se trata al niño, se debe educar a la madre respecto del valor de la luz solar. A menos que sea grave, el raquitismo raramente es una enfermedad fatal por sí misma, aunque el niño puede ser más susceptible a enfermedades infecciosas.
Las deformidades óseas leves tienden a corregirse con el tratamiento, pero en casos más graves puede persistir algún grado de deformidad. Entre las consecuencias más serias están las anormalidades pélvicas que causarán estrechez para el parto y que puede requerir una cesárea.
Osteomalacia
El tratamiento de la osteomalacia es similar al del raquitismo. Se debe suministrar diariamente una dosis de 50 000 UI de vitamina D, como aceite de hígado de bacalao o alguna otra preparación. El calcio se debe dar en lo posible como leche, pero si no existe disponibilidad de leche, en alguna forma medicinal como lactato de calcio.
En mujeres con deformidad pélvica el cuidado prenatal regular es básico; en algunos casos puede ser indispensable la cirugía cesárea antes que el embarazo llegue a término.
PREVENCIÓN
La prevención del raquitismo y la osteomalacia depende de las causas de su incidencia en las comunidades donde se presentan. Casi siempre hay una causa cultural o ambiental que puede ser específica localmente y que puede necesitar una atención particular.
Raquitismo
Se deben tomar medidas para garantizar que todos los niños reciban cantidad adecuada de luz solar. En los climas templados, tales medidas incluyen la erradicación de viviendas insalubres; controlar el humo; construir parques, campos de juego, patios abiertos y jardines; y espacios para los jóvenes.
Los niños deben recibir en su dieta cantidades adecuadas de calcio y vitamina D. La leche y los productos lácteos son especialmente valiosos.
En los casos donde no es posible exponer a los niños a una cantidad correcta de luz solar, se deben suministrar suplementos de vitamina D como el aceite de hígado de bacalao.
Los niños deben concurrir regularmente al servicio de salud de manera que se pueda hacer un diagnóstico precoz de raquitismo para poder tomar las medidas curativas que sean necesarias.
Se debe impartir educación nutricional sobre las necesidades de calcio y vitamina D, y los métodos para obtener cantidades adecuadas de éstos.
Osteomalacia
El cuerpo se debe exponer a una cantidad conveniente de luz solar. Esta necesidad puede estar en conflicto con las costumbres religiosas o culturales, por ejemplo, las que exigen a las mujeres estar ampliamente cubiertas o con velo, o las que prohiben a las mujeres salir de la casa.
Es importante garantizar que se consuma una dieta que contenga cantidades adecuadas de calcio y vitamina D, especialmente las mujeres embarazadas y madres lactantes.
Se deben establecer controles de salud o visitas domiciliarias para poder examinar a las mujeres embarazadas y madres lactantes, y en los casos que sea necesario, suministrarles aceite de hígado de bacalao u otros suplementos de vitamina D. Se debe aconsejar el consumo de alimentos ricos en calcio. Algunas veces se tendrá también que prescribir calcio medicinal (como lactato de calcio).
Se debe impartir educación nutricional e incluir el tema del espaciamiento de los nacimientos.
Niño etíope con raquitismo

Piernas arqueadas en un niño europeo con raquitismo

ENFERMEDAD DE PAGET.
La enfermedad de Paget, la Osteogénesis Imperfecta, el raquitismo y la osteomalacia, son solamente algunos de los trastornos que pueden afectar muy seriamente tanto la calidad como la cantidad de masa ósea en el cuerpo. La Asociación Argentina de Osteología y Metabolismo Mineral, AAOMM, es una especialista en este tipo de males.
Sin dudas, las cifras que indican los niveles de incidencia de la osteoporosis son fuertes: afecta a un tercio de las mujeres de entre 60 y 70 años del mundo, y a los dos tercios de las que tienen más de 80 años, lo que suma a alrededor de 200 millones en todo el mundo. Con estos datos de la Organización Mundial de la Salud (OMS) resulta lógico que sea la enfermedad ósea más popularmente conocida.
Pero, además de ella, hay otras enfermedades, extendidas y con gravedad variable, que afectan al metabolismo del esqueleto en su totalidad o a parte de sus componentes. Son las que profesionalmente se conocen como osteopatías metabólicas o afecciones de los huesos.
“Otras enfermedades que afectan al hueso por algún trastorno de su metabolismo son: enfermedad de Paget, raquitismo y osteomalacia (tiene las mismas causas y características del raquitismo pero se presenta en el adulto) osteogénesis imperfecta, displasia fibrosa, hiperpar atiroidismo primario, osteodistrofia renal”, explica la doctora Diana González, especialista en osteopatías y miembro de la Comisión directiva de la Asociación Argentina de Osteología y Metabolismo Mineral, AAOMM.
La AAOMM es una organización científica fundada en 1983 y dedicada a la investigación y difusión del conocimiento sobre la biología y las enfermedades del esqueleto, especialidad conocida como Osteol ogía.
Forman parte de la Asociación médicos, bioquímicos, biólogos, odontólogos, entre otros profesionales interesados en las enfermedades óseas y el metabolismo mineral.
Los días 24 y 25 de Noviembre se llevó a cabo la XXII Reunión Anual de la Asociación Argentina de Osteología y Metabolismo Mineral, que incluyó conferencias, mesas redondas y ateneos de casos clínicos de interés. También se presentaron los datos preliminares de un importante estudio epidemiológico sobre las fracturas de cadera en la Argentina.
Entre los diversos temas vistos durante la reunión de la AAOMM hay varios directamente vinculados con los conceptos referidos a las osteopatías más diversas:
• Epidemiología de la osteoporosis y de fracturas.
• Aspectos del uso de bifosfonatos en la práctica clínica
• Implantología y uso de biomateriales en odontología.
• Bases moleculares de las enfermedades óseas.
Sin dudas, las cifras que indican los niveles de incidencia de la osteoporosis son fuertes: afecta a un tercio de las mujeres de entre 60 y 70 años del mundo, y a los dos tercios de las que tienen más de 80 años, lo que suma a alrededor de 200 millones en todo el mundo. Con estos datos de la Organización Mundial de la Salud (OMS) resulta lógico que sea la enfermedad ósea más popularmente conocida.
Pero, además de ella, hay otras enfermedades, extendidas y con gravedad variable, que afectan al metabolismo del esqueleto en su totalidad o a parte de sus componentes. Son las que profesionalmente se conocen como osteopatías metabólicas o afecciones de los huesos.
“Otras enfermedades que afectan al hueso por algún trastorno de su metabolismo son: enfermedad de Paget, raquitismo y osteomalacia (tiene las mismas causas y características del raquitismo pero se presenta en el adulto) osteogénesis imperfecta, displasia fibrosa, hiperpar atiroidismo primario, osteodistrofia renal”, explica la doctora Diana González, especialista en osteopatías y miembro de la Comisión directiva de la Asociación Argentina de Osteología y Metabolismo Mineral, AAOMM.
La AAOMM es una organización científica fundada en 1983 y dedicada a la investigación y difusión del conocimiento sobre la biología y las enfermedades del esqueleto, especialidad conocida como Osteol ogía.
Forman parte de la Asociación médicos, bioquímicos, biólogos, odontólogos, entre otros profesionales interesados en las enfermedades óseas y el metabolismo mineral.
Los días 24 y 25 de Noviembre se llevó a cabo la XXII Reunión Anual de la Asociación Argentina de Osteología y Metabolismo Mineral, que incluyó conferencias, mesas redondas y ateneos de casos clínicos de interés. También se presentaron los datos preliminares de un importante estudio epidemiológico sobre las fracturas de cadera en la Argentina.
Entre los diversos temas vistos durante la reunión de la AAOMM hay varios directamente vinculados con los conceptos referidos a las osteopatías más diversas:
• Epidemiología de la osteoporosis y de fracturas.
• Aspectos del uso de bifosfonatos en la práctica clínica
• Implantología y uso de biomateriales en odontología.
• Bases moleculares de las enfermedades óseas.
• Uso de células mesenquimáticas osteo progenitoras en la reparación de tejidos: ¿un nuevo concepto terapéutico?
Enfermedad de Paget
No es un trastorno generalizado como sí lo es la osteoporosis: mientras que esta última afecta a todos los huesos del cuerpo, la enfermedad de Paget se mantiene usualmente localizada en un área, afectando a uno o a varios huesos. Es una enfermedad crónica que produce una excesiva destrucción y regeneración en el tejido óseo, cuyo resultado es un hueso más frágil, que puede doler, deformarse y fracturarse.
Aun cuando se desconoce todavía con certeza cuáles son las causas de la enfermedad de Pager, se sabe que hay una predisposición familiar a padecerla.
Diversos estudios indican que los familiares de una persona con enfermedad de Paget poseen siete veces más peligro de desarrollar la enfermedad que una persona que no tiene familiares afectados.
Sólo en los Estados Unidos, se estima que el 1,3 por ciento de los hombres y mujeres cuyas edades rondan entre los 45 y 74 años padecen el mal, con una presencia dos veces más fuerte entre el sexo masculino.
Muchos pacientes ignoran que tienen la enfermedad de Pager, debido a que un elevado porcentaje de los casos se presenta (a simple vista) como asintomática.
El dolor óseo, si bien tiene características particulares, se puede confundir con el dolor originado en la artrosis u otras enfermedades. En algunos casos, el diagnóstico ocurre sólo cuando se han presentado complicaciones: por ejemplo una fractura, artrosis de una articulación vecina al hueso afectado de Paget, o simplemente porque en un análisis de laboratorio surge un valor anormalmente alto de fosfatasa alcalina.
Para diagnosticar la enfermedad, además de las radiografías simples, hay otros estudios: análisis del nivel de fosfatasa alcalina (total y la específica del hueso) en la sangre, centellograma óseo y tomografía del hueso. El pronóstico para las personas diagnosticadas con enfermedad de Paget es generalmente bueno, “Si bien la enfermedad de Paget hasta hoy no se cura con ninguna medicación disponible –puntualiza la doctora González- desde hace aproximadamente 30 años, disponemos en nuestro país de un tipo de droga, los bifosfonatos, con los cuales se puede controlar completamente la enfermedad, evitando su progresión y complicaciones.”
Los bifosfonatos reducen el número de osteoclastos y su actividad, disminuyendo al mismo tiempo por este mecanismo la destrucción ósea exagerada que caracteriza a esta enfermedad.
Por este motivo también se utilizan en oncología cuando hay un compromiso óseo provocado por un tumor o metástasis.
Miembros fundadores de la AAOMM, como los Dres. Gunther Fromm y Carlos Mautalen han aportado una extensa y valiosa experiencia con el uso de estas drogas en el tratamiento de la enfermedad de Paget.
Osteogénesis Imperfecta
La OI significa que hay huesos formados imperfectamente debido a defectos genéticos. El colágeno es la principal proteína del tejido conectivo del cuerpo y es la estructura sobre la cual se forman los huesos y tejidos. Si dicha estructura de colágeno es defectuosa, los huesos se fracturan con facilidad, la piel es torna delgada y transparente y los músculos disminuyen su tonicidad.
Es allí cuando se habla de osteogénesis imperfecta. Las personas con OI tienen menos colágeno de lo normal, y si lo poseen en la cantidad necesaria, el mismo es de mala calidad.
Existen por lo menos cuatro tipos de OI, llamados tipo I, II, III y IV, y las características de uno a otro y de persona a persona varían bastante, Es la enfermedad de los “huesos de cristal” porque en general, se fracturan fácilmente.
Además los afectados por esta enfermedad pueden tener baja estatura, pérdida del sentido de la audición, dientes descoloridos y débiles, deformidades esqueléticas en las extremidades producidas como consecuencia de las múltiples fracturas, escoliosis (curvatura de la espina dorsal). “Actualmente no hay una cura disponible para la Osteogénesis Imperfecta.
El tratamiento busca, básicamente, prevenir o evitar las fracturas, que son el síntoma excluyente, aumentando la masa ósea y la fortaleza de los múscul os”, explica la doctora Diana González, desde la AAOMM.
Hiperparatiroidismo primario
Es un problema hormonal debido a que una o más de las glándulas paratiroideas (son cuatro, y están ubicadas en el cuello, cerca de la glándula tiroidea) producen demasiada hormona paratiroidea (PTH). En condiciones normales, esta hormona es responsable de mantener un correcto balance de calcio en el organismo. Pero si su producción resulta siendo exagerada, como ocurre cuando hay un tumor de paratiroides, el calcio en la sangre puede aumentar excesivamente porque es removido del hueso. El resultado es también una descalcificación de grado variable. La incidencia de esta enfermedad en mujeres es tres veces más alta que en los hombres, y aumenta con la edad
Muchos pacientes no presentan síntomas, y el trastorno se detecta frecuentemente cuando la persona es evaluada para saber si tiene una osteoporosis: pueden aparecer cambios en algunos valores de laboratorio, o una densitometría disminuida. Pérdida del apetito, sed, letargo, fatiga, debilidad muscular, dolor articular, constipación, necesidad frecuente de orinar son algunos de los síntomas, que empeoran cuando el calcio en sangre se elevada excesivamente.
La doctora González explica que el tratamiento en general es quirúrgico, con lo que se resuelve completamente el problema en la mayoría de los casos. Además, hay opciones de tratamiento con bifosfonatos o tratamiento hormonal con estrógenos, cuando la circunstancia del paciente impide el abordaje quirúrgico.
Acerca de la AAOMM
La AAOMM es una organización científica fundada en 1983 dedicada a la investigación y difusión del conocimiento sobre la biología y las enfermedades del esqueleto, especialidad conocida como Osteología. La conforman médicos, bioquímicos, biólogos, odontólogos y otros los profesionales interesados en las enfermedades óseas y el metabolismo mineral. Entre sus actividades académicas figuran la organización periódica de reuniones científicas, mesas redondas y la edición de una revista científica, con referato, que publicará trabajos de investigación originales. Actualmente, la AAOMM está afiliada a la International Osteoporosis Foundation (IOF) y a la Sociedad Iberoamericana de Osteología y Metabolismo Mineral (SIBOMM). Para mayor información, favor de visitar la página www.aaomm.org.ar
La AAOMM es una organización científica fundada en 1983 dedicada a la investigación y difusión del conocimiento sobre la biología y las enfermedades del esqueleto, especialidad conocida como Osteología. La conforman médicos, bioquímicos, biólogos, odontólogos y otros los profesionales interesados en las enfermedades óseas y el metabolismo mineral. Entre sus actividades académicas figuran la organización periódica de reuniones científicas, mesas redondas y la edición de una revista científica, con referato, que publicará trabajos de investigación originales. Actualmente, la AAOMM está afiliada a la International Osteoporosis Foundation (IOF) y a la Sociedad Iberoamericana de Osteología y Metabolismo Mineral (SIBOMM). Para mayor información, favor de visitar la página www.aaomm.org.ar
OSTEOPOROSIS
La osteoporosis es una disminución de la masa ósea y de su resistencia mecánica que ocasiona susceptibilidad para las fracturas. Es la principal causa de fracturas óseas en mujeres después de la menopausia y ancianos en general. La osteoporosis no tiene un comienzo bien definido y, hasta hace poco, el primer signo visible de la enfermedad acostumbraba a ser una fractura de la cadera, la muñeca o de los cuerpos vertebrales que originaban dolor o deformidad.
La menopausia es la principal causa de osteoporosis en las mujeres, debido a disminución de los niveles de estrógenos. La pérdida de estrógenos por la menopausia fisiológica o por la extirpación quirúrgica de los ovarios, ocasiona una rápida pérdida de hueso. Las mujeres, especialmente las caucásicas y asiáticas, tienen una menor masa ósea que los hombres. La pérdida de hueso ocasiona una menor resistencia del mismo, que conduce fácilmente a fracturas de la muñeca, columna y la cadera.

Vertebras con disminución en la cantidad de hueso (zonas más oscuras). Tomado de la Universidad de Utah
Una mayor probabilidad de desarrollar osteoporosis se relaciona con:
Menopausia precoz, natural o quirúrgica
Consumo del alcohol o cafeína
Tabaquismo
Períodos de amenorrea
Algunos medicamentos como el uso prolongado de córticoesteroides
Procesos como enfermedad tiroidea, artritis reumatoide y problemas que bloquean la absorción intestinal de cálcio
Dieta pobre en cálcio por períodos prolongados, especialmente durante la adolescencia y la juventud
Vida sedentaria
En situaciones de menopausia precoz, las mujeres deben tomar estrógenos para prevenir la pérdida post-menopáusica de hueso; se debe de añadir un progestágeno si el útero está intacto. El reemplazamiento estrogénico es un tratamiento efectivo para prevenir la pérdida post-menopáusica de hueso y es también efectivo en la prevención de fracturas osteoporóticas. El tratamiento hormonal sustitutivo requiere un estricto control ginecológico y una cuidadosa selección de pacientes.
Las mujeres post-menopáusicas con baja masa ósea o osteoporosis establecida y que tengan contraindicación para el tratamiento hormonal sustitutivo, los Bifosfonatos (Alendronato o Etidronato) y la Calcitonina, son medicamentos efectivos para prevenir la pérdida de hueso.
El caminar y los ejercicios de extensión de la columna pueden estabilizar o incluso incrementar ligeramente la masa ósea y mejorar el balance y la fuerza musculares, previniendo caídas y fracturas.
Las fracturas vertebrales deben tratarse inicialmente con reposo, analgésicos, lumbostato y rehabilitacion. Otros posibles tratamientos, actualmente en estudio, incluyen vitamina D, fluoruros y hormona paratiroidea.
osteogénesis imperfecta
¿CUAL ES SU CAUSA?
Los huesos están sometidos a un remodelado contínuo mediante procesos de formación y reabsorción, y también sirven como reservorio de calcio del organismo. A partir de los 35 años se incia la pérdida de pequeñas cantidades de hueso. Múltiples enfermedades o hábitos de vida pueden incrementar la pérdida de hueso ocasionando osteoporosis a una edad más precoz. Algunas mujeres están, también, predispuestas a la osteoporosis por una baja masa ósea en la edad adulta.La menopausia es la principal causa de osteoporosis en las mujeres, debido a disminución de los niveles de estrógenos. La pérdida de estrógenos por la menopausia fisiológica o por la extirpación quirúrgica de los ovarios, ocasiona una rápida pérdida de hueso. Las mujeres, especialmente las caucásicas y asiáticas, tienen una menor masa ósea que los hombres. La pérdida de hueso ocasiona una menor resistencia del mismo, que conduce fácilmente a fracturas de la muñeca, columna y la cadera.
Vertebras con disminución en la cantidad de hueso (zonas más oscuras). Tomado de la Universidad de Utah
Una mayor probabilidad de desarrollar osteoporosis se relaciona con:
IMPACTO EN LA POBLACIÓN
La osteoporosis afecta a una de cada cinco mujeres de más de 45 años y a cuatro de cada diez de más de 75.DIAGNÓSTICO
Se puede medir la masa ósea, y por tanto su disminución en el adulto, con técnicas de densitometría o de tomografía computadorizada cuantitativa.TRATAMIENTO
El mejor tratamiento de la osteoporosis es la prevención. Una ingesta adecuada de cálcio y el ejercicio físico durante la adolescencia y la juventud, puede incrementar el pico de masa ósea, lo cual redunda en una reducción de la pérdida de hueso y en un menor riesgo de fractura en años posteriores. El consumo adecuado de cálcio y de vitaminas durante la madurez es esencial para la salud del hueso.En situaciones de menopausia precoz, las mujeres deben tomar estrógenos para prevenir la pérdida post-menopáusica de hueso; se debe de añadir un progestágeno si el útero está intacto. El reemplazamiento estrogénico es un tratamiento efectivo para prevenir la pérdida post-menopáusica de hueso y es también efectivo en la prevención de fracturas osteoporóticas. El tratamiento hormonal sustitutivo requiere un estricto control ginecológico y una cuidadosa selección de pacientes.
Las mujeres post-menopáusicas con baja masa ósea o osteoporosis establecida y que tengan contraindicación para el tratamiento hormonal sustitutivo, los Bifosfonatos (Alendronato o Etidronato) y la Calcitonina, son medicamentos efectivos para prevenir la pérdida de hueso.
El caminar y los ejercicios de extensión de la columna pueden estabilizar o incluso incrementar ligeramente la masa ósea y mejorar el balance y la fuerza musculares, previniendo caídas y fracturas.
Las fracturas vertebrales deben tratarse inicialmente con reposo, analgésicos, lumbostato y rehabilitacion. Otros posibles tratamientos, actualmente en estudio, incluyen vitamina D, fluoruros y hormona paratiroidea.
EL PAPEL DEL REUMATÓLOGO EN EL TRATAMIENTO DE LA OSTEOPOROSIS
Como especialista entrenado para evaluar enfermedades reumáticas, los Reumatólogos pueden diferenciar la osteoporosis de las enfermedades que originan pérdida de hueso y establecer y monitorizar un programa terapéutico. Los Reumatólogos son activos agentes en la educación del público en general y de otros médicos, sobre este grave problema sanitario.La osteogénesis imperfecta es una enfermedad congénita que se caracteriza porque los huesos de las personas que la sufren se rompen muy fácilmente, con frecuencia tras un traumatismo mínimo e incluso sin causa aparente.
Se conocen varios tipos de la enfermedad, y su variación es muy grande de un individuo a otro. Incluso dentro del mismo tipo, puede haber personas con una mayor o menor impregnación.
Por decirlo con un ejemplo práctico, algunos pacientes sufren diez fracturas a lo largo de su vida, en tanto que otros pueden llegar a tener varios cientos de ellas.
...
La osteogénesis imperfecta se debe a la insuficiente y/o defectuosa formación del colágeno del cuerpo, como consecuencia de un fallo genético. El colágeno es una proteína de los tejidos y su función en la formación de los huesos se puede comparar a la de las nervaduras metálicas en torno a las cuales se monta la estructura de hormigón de una viga. Si la nervadura no es fuerte o no existe, la pieza de hormigón no adquirirá la forma adecuada o será sumamente frágil.
La OI no se puede paliar, por tanto, suministrando calcio a los enfermos. No existe hasta el presente ninguna forma de inducir a las células del cuerpo a producir más colágeno o producir colágeno de calidad.
...
La osteogénesis imperfecta tiene un pronóstico muy variable, dependiendo del grado en que cada individuo esté afectado. La enfermedad en sí no es letal. Sin embargo, las personas afectadas por las formas más graves pueden tener importantes problemas colaterales.
Mientras que los afectados por tipos más leves (I y IV según la clasificación de Sillence), en general no tienen más complicaciones que las que impongan sus fracturas y deformaciones óseas, así como las intervenciones quirúrgicas que sean necesarias para tratarlas, la OI del tipo II (siempre según Sillence) suele revestir mucha gravedad y puede llegar a ser letal, debido a las hemorragias que causan las fracturas múltiples en el recién nacido.
El tipo III de Sillence tiene un pronóstico variable. En los casos en que la deformación ósea es grave y el volumen torácico es escaso, se pueden presentar problemas de ventilación, que en algunos casos pueden dar lugar a neumonías.
En todos los tipos se pueden presentar también problemas cardiovasculares de diferente pronóstico.
A pesar de la deformación ósea y la frecuencia de fracturas, la longevidad de una persona afectada por OI es igual a la de cualquier otra. Sus cualidades intelectuales no están mermadas de ninguna forma por la enfermedad y pueden llevar una vida normal, dentro de las limitaciones que imponga el grado de movilidad de cada uno.
...
Es difícil determinar el número de personas afectadas por la enfermedad, ya que no hay censos donde consten semejantes datos. Además, podemos partir de la base de que hay gran cantidad de afectados con síntomas muy leves que no han sido ni serán nunca diagnosticados.
Yo he leído estimaciones que dicen que la incidencia es de uno entre veinte o treinta mil nacimientos (OIF), y otras según las cuales la cifra debe de andar por un afectado entre cada diez mil nacidos, incluyendo los casos que son diagnosticados a posteriori.
En cuanto a la forma en que se transmite, los especialistas no acaban de ponerse de acuerdo. Hasta no hace mucho parecía claro que la forma de transmisión de los tipos I y IV era dominante (estos dos tipos se pueden estudiar en familias donde la enfermedad aparece a lo largo de generaciones). La opinión generalmente aceptada era hasta hace muy poco que tanto el tipo II como el tipo III son el resultado de una transmisión recesiva, pero los más recientes estudios parecen reflejar dudas fundadas sobre esta generalización y apuntan a la posibilidad de que el mosaicismo (error genético en las células germinales de los progenitores) desempeñe un papel importante en la transmisión de los casos más graves de OI.
Además, la enfermedad también puede ser el resultado de una mutación espontánea y aparecer en familias sin ningún antecedente. En algunos casos, lo que puede parecer una mutación espontánea no lo es: hay afectados con síntomas tan leves que ignoran su condición hasta que tienen hijos con la enfermedad.
En cualquier caso, está claro que los afectados por osteogénesis imperfecta tienen un cincuenta por ciento de probabilidades de transmitirla a su descendencia.
Introducción
La cuestión de la terapia es muy conflictiva porque cada caso es muy particular (me atrevo a decir que no hay dos pacientes con osteogénesis imperfecta cuyos historiales sean idénticos) y cada cual habla de la feria según como le fue en ella. Lo que pueden leer a continuación son hechos de los que me he ido informando a lo largo de estos años. En algunos aspectos, me refiero a mi experiencia, que no es ni mejor ni peor que otras, y sí muy particular mía. Repito que no tengo formación médica y que no excluyo meteduras de pata por mi parte cuando me refiera a términos especializados. En cualquier caso, lo que voy a contar ahora es simple información general. Cualquiera que estuviera interesado en ella, en lugar de creer a ciegas lo que yo digo, debería consultarlo con un buen especialista (que los hay).
Si entendemos como terapia un tratamiento que cure la enfermedad, la respuesta es no. El problema de las personas que sufren osteogénesis imperfecta es que las células de su organismo no producen una cantidad suficiente de colágeno (o el que producen es de calidad defectuosa). Hasta ahora no se ha hallado ningún método para inducir a las células a mejorar o incrementar su producción de colágeno.
Según me comentó el pediatra de mi hijo, se ha logrado, en experimentos «in vitro», inducir a una célula a producir colágeno. La dificultad de conseguir este mismo resultado a escala humana está al alcance de la mano: ¿cómo inducir atodas las células del organismo a producir mejor colágeno y en mayor cantidad?
Según me comentó el pediatra de mi hijo, se ha logrado, en experimentos «in vitro», inducir a una célula a producir colágeno. La dificultad de conseguir este mismo resultado a escala humana está al alcance de la mano: ¿cómo inducir atodas las células del organismo a producir mejor colágeno y en mayor cantidad?
Así las cosas, cuando hablamos de terapia de la osteogénesis imperfecta nos estamos refiriendo a tratamientos correctivos y paliativos de la enfermedad, no a una curación.
En este sentido, hay tres aspectos terapéuticos interesantes: la fisioterapia, el correcto tratamiento de las fracturas y la introducción de agujas telescópicas Bailey en los huesos largos para prevenir curvaturas y fracturas.
Mención aparte merece el empleo de diferentes substancias que ayudan a incrementar la densidad ósea, bisfosfonatos y pamidronato. Durante estos últimos años se están llevando a cabo en todo el mundo experimentos en este campo y los resultados parecen ser muy prometedores.
1. Clasificación del Dr. SillenceEl Dr. Sillence es una de las mayores autoridades mundiales en materia de OI. Si no me equivoco mucho, vive y trabaja en Australia. El estableció hace ya algunos años una clasificación de la osteogénesis imperfecta en cuatro tipos que se ha convertido en el punto obligado de referencia de otros especialistasTipo IEs el tipo más frecuente de OI. Se transmite genéticamente como autosomal dominante, pero también puede ser el resultado de una mutación espontánea. Como promedio se puede decir que los individuos con tipo I pueden tener de veinte a treinta fracturas antes de la pubertad, pero pueden ser más y también menos. La incidencia de fracturación se reduce después de la pubertad. En determinados casos es posible realizar un diagnóstico prenatal de estos tipos. Las mujeres adultas con tipo I de OI suelen volver a tener fracturas tras la menopausia. Con todo, el tipo I de OI puede considerarse como leve.Las personas con tipo I pueden presentar una o más de las siguientes características:fragilidad ósea
Tipo IIAproximadamente el 10 por ciento de las personas afectadas por la OI son del tipo dos, que resulta de una nueva mutación y es la forma más severa de OI. Los niños que nacen con este tipo de OI presentan fracturas perinatales, miembros poco desarrollados y curvos, y tienen los huesos extremadamente frágiles. Con frecuencia fallecen poco después de nacer (una de las razones de esta mortalidad temprana son las hemorragias internas que se producen como consecuencia de las numerosas fracturas) El diagnóstico prenatal es posible: en la ecografía se pueden apreciar la curvatura de los miembros y determinadas fracturas.
Con todo, las clasificaciones no siempre son exactas. Hay gente que comparte características de los tipos III/IV, otros que se definen como II/III, y otros que simplemente quedan fuera. Hay estudios sobre familias con dentinogénesis imperfecta, escleróticas azules y baja estatura, pero con ausencia de fracturas, a los que cuesta incluir en el tipo I.Tipo IIIAproximadamente el 20 por ciento de las personas con OI tienen el tipo III. Estos enfermos sufren con frecuencia fracuras espontáneas. No es infrecuente encontrar pacientes del tipo III que hayan sufrido más de veinte fracturas durante los tres primeros años de vida. Al llegar a la pubertad, el número puede haber ascendido a más de cien. Se presentan con mucha frecuencia articulaciones hiperextensibles y un desarrollo muscular pobre. El diagnóstico prenatal puede ser posible mediante ecografía.
El pronóstico de la osteogénesis imperfecta tipo III es severo. Debido a la curvatura de las extremidades inferiores y a su fragilidad, la mayoría de los afectados por este tipo no puede caminar.Los afectados con el tipo III pueden presentar una o más de las siguientes características:
Tipo IVEl pronóstico de la osteogénesis imperfecta tipo IV va de leve a moderado. La mayoría de las fracturas se presentan durante la infancia, pero también hay recurrencia en el caso de mujeres menopaúsicas. La fragilidad ósea de los afectados por este tipo se pone con frecuencia de manifiesto a través de la curvatura de los huesos largos, especialmente los huesos de las piernas.Las personas con tipo IV pueden presentar una o más de las siguientes características:
Tengo que añadir que conozco a un par de personas con OI que manifiestan claramente que no les interesa en absoluto saber a qué grupo pertenecen. Dicen que esas cosas le importan al médico: al paciente le dan lo mismo.
La osteogénesis imperfecta es una enfermedad conocida desde la antigüedad (se han descubierto momias egipcias de personas afectadas por la OI).
Tradicionalmente se hablaba de dos tipos de OI:Osteogénesis imperfecta congénita.Esta clasificación englobaba a los tipos de OI que eran detectados inmediatamente después del nacimiento, debido a las deformaciones y las fracturas visibles. En este tipo se incluía la forma letal (el tipo II de Sillence) y las formas severas (tipo III de Sillence).
Osteogénesis imperfecta tarda.Con este nombre se referían los médicos a las formas de OI que se detectaban posteriormente debido a la frecuencia de fracturación y a las deformaciones en los miembros. Esta forma incluía a los tipos I y IV de la clasificación de Sillence.Si bien esta clasificación está más que superada por la del Dr. Sillence, mucho más clara y efectiva, aún de puede encontrar en muchos manuales de Medicina, e incluso en artículos relativamente modernos. De hecho, el primer diagnóstico que se le hizo a mi hijo fue precisamente «osteogénesis imperfecta tarda».
El equipo del Dr. Glorieux, del Hospital Shriners de Montreal, ha definido nuevos tipos de OI, más allá de los cuatro de Sillence.Se habla además de otros subgrupos del tipo IV con características comunes, que podrían definirse como grupos independientes.
El grupo IV de Sillence era el más heterogéneo, con una gama de características mucho más variadas. Dentro de este grupo, Glorieux ha detectado un subgrupo con características comunes. Entre ellas, la formación de callo hipertrófico en las zonas de fractura o de osteotomía. Más información (en inglés) sobre este nuevo grupo de OI se puede encontrar enTipo V OI.
En la actualidad, el Dr. Horacio Plotkin trabaja en la esquematización de una nueva clasificación de la OI, más acorde al diagnóstico clínico. La variedad de la osteogénesis imperfecta es tal que no cabe duda de que sera muy difícil encontrar unas categorías que tengan carácter definitivo.
Se ha hablado siempre de una transmisión de la osteogénesis imperfecta según un modelo autosómico dominante, con excepciones en presentaciones muy graves de la enfermedad que supuestamente responden a modelos de transmisión recesiva. Al respecto tienen ustedes a continuación dos artículos informativos aparecidos en la lista estadounidense OI-Parents, fundada y coordinada por padres de niños con osteogénesis imperfecta:
Líneas de investigación y tratamiento pasadas y presentes
Las personas con osteogénesis imperfecta tienen un cincuenta por ciento de probabilidades de que sus hijos biológicos hereden la enfermedad. Además, la OI se presenta como mutación espontánea o como resultado de mutaciones en las células germinales de los padres. Muchos futuros padres que saben que sus hijos pueden tener OI están interesados en contar con métodos de diagnóstico que ayuden a determinar si el hijo no nacido va a tener o no la enfermedad. También hay padres que ya han perdido a un hijo a causa de una osteogénesis imperfecta grave y desean saber si existen métodos para determinar antes de que el niño nazca si va a estar afectado o no.Para su información he recopilado y traducido información sobre diagnóstico prenatal de la osteogénesis imperfecta. Las notas proceden en su mayoría de mensajes recibidos a través de las listas de osteogénesis imperfecta en inglés. Los autores de los mensajes son médicos genetistas y afectados que se han sometido a las diversas pruebas de diagnóstico prenatal que se especifican a continuación.
Hipofosfatasia
Hay una heterogeneidad notable en la presentación de los síntomas dependiendo en gran medida de la edad en la presentación inicial, que van desde la muerte en el útero a problemas relativamente simples, con dentadura en la vida adulta.
Aunque varios subtipos clínicos de la enfermedad se ha caracterizado sobre la base de la edad en que se descubren lesiones en el esqueleto, la enfermedad se entiende mejor como un único espectro continuo de gravedad.
En el período perinatal, hipofosfatasia es la forma más perniciosa de hipofosfatemia. Se expresa en el útero como hipomineralización profunda que se traduce en membraneceum cápita, deformado o reducido las extremidades durante la gestación y el nacimiento y la muerte rápida debido a la insuficiencia respiratoria.
Muerte del feto no es infrecuente y supervivencia a largo plazo es poco común.
Los recién nacidos que logran sobrevivir a varios días o semanas sufren cada vez mayor compromiso respiratorio debido a la enfermedad del pecho raquítico y pulmones hipoplásicos, y en última instancia, la insuficiencia respiratoria.
Epilepsia (ataques) pueden ocurrir y resultar letal (vide infra).
Osteoide excesiva puede invadir el espacio de la médula y provocar anemia mielotísica.
En los exámenes radiográficos, hipofosfatasia perinatal se distingue fácilmente de incluso las formas más graves de la osteogénesis imperfecta y el enanismo congénito.
Algunos esqueletos de muertos casi no muestran la mineralización, mientras que otros han marcado undermineralization óseas y severos cambios raquíticos, de vez en cuando, puede haber ausencia peculiar total o parcial de osificación en una o más vértebras.
En el cráneo, los huesos membranosos individuales pueden calcificarse sólo en sus centros, lo que las áreas de la calvaruim osificar la ilusión de que las suturas del cráneo están muy separadas, cuando en realidad son funcionalmente cerrado.
Otra característica radiográfica es inusual espolones óseos que sobresalen lateralmente de la midshafts del cúbito y fíbulas. A pesar de la considerable de paciente a paciente variabilidad y la diversidad de los hallazgos radiológicos, la radiografía puede considerarse diagnóstico.
El subtipo infantil se describe con regalos hipofosfatasia en los primeros 6 meses de vida. El desarrollo postnatal a menudo parece normal hasta el inicio de la mala alimentación y aumento de peso insuficiente, y las manifestaciones clínicas del raquitismo son reconocidos.
A pesar de las suturas del cráneo parecen ser amplio, esto refleja hipomineralización del cráneo, y hay a menudo "funcional" craneosinostosis, y si el paciente sobrevive la infancia, estas suturas pueden fusionarse de manera permanente.
A menudo, un tórax batiente de fracturas de costillas, deformidad costal, etc conduce al compromiso respiratorio y neumonía. Hipercalcemia y hypercalcenuria también son comunes y pueden explicar la nefrocalcinosis, compromiso renal y episodios de vómitos recurrentes.
Las características radiográficas son sorprendentes aunque por lo general menos graves que las que se encuentran en la hipofosfatasia perinatal.
En algunos pacientes con diagnóstico reciente una transición abrupta de relativamente de apariencia normal diáfisis de metáfisis no calcificado aparece, lo que sugiere la aparición de cambios metabólicos bruscos.
Además, los estudios de serie de la radiografía puede mostrar la persistencia de la mineralización ósea disminuida (es decir, el raquitismo) y revelar la desmineralización generalizada gradual.
La mortalidad se estima en un 50% en el primer año de vida.
En la infancia, la expresión clínica hipofosfatasia es extremadamente variable. Como resultado de aplasia, hipoplasia o displasia del cemento dental, pérdida prematura de dientes temporales (es decir, antes de la edad de 5 años) se produce.
Con frecuencia, los incisivos se desprenden primera vez en casi toda la dentición temporal se exfolia antes de tiempo. Las radiografías dentales a veces muestran las cámaras de pulpa agrandadas y canales de la raíz característica de los "dientes de cáscara" del raquitismo.
Los pacientes también pueden experimentar retraso en caminar, un andar característico de pato, se quejan de la rigidez y el dolor, y tienen una debilidad muscular apendicular (especialmente en los muslos), en consonancia con miopatía progresiva.
Por lo general, las radiografías muestran deformidades raquíticas y característicos defectos óseos cerca de los extremos de los principales huesos largos (es decir, "lenguas" de la radio-que sobresalen de la placa de crecimiento raquítico en el metaphsysis).
Retraso del crecimiento, fracturas frecuentes y la osteopenia son comunes.
En los bebés afectados y los niños pequeños no es raro que, a pesar de la apariencia de muy "abierto" fontanelas en los estudios radiográficos, para la sinostosis funcionales de las suturas craneales que se produzca.
La ilusión de "abrir" los resultados de las fontanelas amplias zonas de la bóveda craneal hipomineralizada. Posteriormente verdadera fusión prematura de las suturas craneales óseas pueden elevar la presión intracraneal.
En edad adulta, pueden presentar Hipofosfatasia durante la mediana edad. Con frecuencia, hay una historia de raquitismo, pérdida prematura de dientes de leche, o la pérdida temprana de la dentición adulta seguida de una salud relativamente buena.
Los hallazgos dentales
A menudo, uno de los primeros síntomas de hipofosfatemia es la pérdida temprana de hoja caduca (el bebé o dientes primarios) con la raíz intacta.
Los investigadores han documentado recientemente una correlación positiva de anomalías dentales con el fenotipo clínico.
La mala dentadura se observa en los adultos.
Laboratorio de Ensayo
El síntoma patognomónico es la actividad subnormal séricos de fosfatasa alcalina (ALP). En general, la gravedad clínica refleja el grado de deficiencia de la enzima.
El marcador de sustrato más sensible para la hipofosfatasia es un aumento de piridoxal 5-fosfato (PLP) el nivel de plasma, que a menudo se correlaciona con la severidad de la enfermedad. Y, aunque sigue siendo sólo una técnica de investigación, la cuantificación del pirofosfato inorgánico urinario (PPi) niveles, que son elevados en la mayoría de los pacientes hipofosfatasia, se ha reportado para detectar con precisión los transportistas.
Además, el aumento de los niveles urinarios de fosfoetanolamina (PEA) se observan en la mayoría de los pacientes. Disponibilidad de la prueba de fosfatasa alcalina en suero ajustado por edad está extendida y se incluye en muchos CHEM20 paneles.
Radiografía
A pesar de paciente a paciente variabilidad y la diversidad de los hallazgos radiológicos, la radiografía es diagnóstica en hipofosfatasia infantil, y puede revelar las anormalidades características en otras formas.
Evidencia radiológica de los defectos óseos se encuentran en casi todos los pacientes e incluye hipomineralización, cambios raquíticos, osificación incompleta de vertebrados y, en ocasiones, los espolones óseos en el lateral del cúbito y fíbulas. Disponibilidad de Rayos X se han generalizado.
En los recién nacidos rayos X fácilmente distinguir HPP perinatal incluso de las formas más graves de la osteogénesis imperfecta y el enanismo congénito.
Algunos esqueletos de muertos casi no muestran la mineralización, mientras que otros han marcado undermineralization óseas y severos cambios raquíticos, y de vez en cuando, puede haber ausencia peculiar total o parcial de osificación en una o más vértebras.
En el cráneo, los huesos membranosos individuales pueden calcificarse sólo en sus centros, lo que parece que las áreas de la bóveda craneal osificar tiene suturas craneales que están muy separadas, cuando en realidad son funcionalmente cerrado.
Las lenguas de los radio-frecuencia sobresalen de la metáfisis en el eje del hueso.
En los bebés, características radiográficas de la HPP infantiles son sorprendentes aunque por lo general menos graves que las que se encuentran en la HPP perinatal.
En algunos pacientes recién diagnosticados, una transición abrupta de relativamente de apariencia normal diáfisis de metáfisis sin calcificar parece sugerir un cambio metabólico abrupta se ha producido.
Estudios de serie de la radiografía puede revelar la persistencia de la mineralización ósea disminuida (es decir, el raquitismo), casos de la esclerosis y la desmineralización generalizada gradual.
En los adultos, los rayos X pueden revelar seudofracturas bilateral de fémur en la diáfisis subtrocantéreas lateral. Estos psuedofractures pueden permanecer por años o empeorar, pero no puede sanar hasta que se rompen por completo o si el paciente recibe la fijación intramedular. Estos pacientes también pueden experimentar recurrentes fracturas del hueso metatarsiano.
El análisis genético
Todos los subtipos clínicos de hipofosfatasia se han atribuido a mutaciones genéticas en el gen que codifica TNSALP, que se localiza en el cromosoma 1p36.1-34 en los seres humanos (ALPL; OMIM # 171 760).
Aproximadamente 204 mutaciones diferentes han sido descritas en el gen TNSALP. Una lista actualizada de mutaciones en línea en http://www.sesep.uvsq.fr/database_hypo/Mutation.html. Alrededor del 80% de las mutaciones son mutaciones sin sentido.
El número y la diversidad de los resultados de las mutaciones en la expresión fenotípica muy variable. Parece haber una correlación entre genotipo y fenotipo en la hipofosfatasia ".
Análisis de la mutación es posible y está disponible en tres laboratorios
Hay una heterogeneidad notable en la presentación de los síntomas dependiendo en gran medida de la edad en la presentación inicial, que van desde la muerte en el útero a problemas relativamente simples con la dentición en la vida adulta.
Aunque varios subtipos clínicos de la enfermedad se ha caracterizado sobre la base de la edad en que se descubren lesiones en el esqueleto, la enfermedad se entiende mejor como un solo espectro continuo de gravedad.
En el período perinatal, hipofosfatasia es la forma más perniciosa de hipofosfatemia. Se expresa en el útero como hipomineralización profunda que se traduce en membraneceum cápita, deformado o reducido las extremidades durante la gestación y el nacimiento y la muerte rápida debido a la insuficiencia respiratoria.
Muerte del feto no es infrecuente y supervivencia a largo plazo es poco común.
Los recién nacidos que sobreviven varios días o semanas sufren cada vez mayor compromiso respiratorio debido a la enfermedad del pecho raquítico y pulmones hipoplásicos, y en última instancia, la insuficiencia respiratoria.
La epilepsia (ataques) pueden ocurrir y resultar letal (vide infra).
Osteoide excesiva puede invadir el espacio de la médula y provocar anemia mielotísica.
En los exámenes radiográficos, hipofosfatasia perinatal se distingue fácilmente de incluso las formas más graves de la osteogénesis imperfecta congénita y enanismo .
Algunos esqueletos de muertos casi no muestran la mineralización, mientras que otros han marcado undermineralization óseas y severos cambios raquíticos, de vez en cuando, puede haber ausencia peculiar total o parcial de la osificación en una o más vértebras.
En el cráneo, los huesos membranosos individuales pueden calcificarse sólo en sus centros, dando a las áreas de la calvaruim osificar la ilusión de que las suturas del cráneo están muy separadas, cuando en realidad son funcionalmente cerrado.
Otra característica radiográfica es inusual espolones óseos que sobresalen lateralmente de la midshafts del cúbito y fíbulas. A pesar de la considerable de paciente a paciente variabilidad y la diversidad de los hallazgos radiológicos, los rayos X puede considerarse diagnóstico.
El subtipo infantil se describe con regalos hipofosfatasia en los primeros 6 meses de vida. El desarrollo postnatal a menudo parece normal hasta el inicio de la mala alimentación y aumento de peso insuficiente, y las manifestaciones clínicas de raquitismo son reconocidos.
A pesar de las suturas craneales parecen estar bien, esto refleja hipomineralización del cráneo, y con frecuencia hay "funcional" craneosinostosis, y si el paciente sobrevive la infancia, estas suturas pueden fusionarse de manera permanente.
A menudo, un tórax inestable de fracturas de costillas, deformidad costal, etc lleva a compromiso respiratorio y neumonía . Hipercalcemia y hypercalcenuria también son comunes y pueden explicar la nefrocalcinosis, compromiso renal, y los episodios recurrentes de vómitos .
Las características radiográficas son sorprendentes aunque por lo general menos graves que las que se encuentran en la hipofosfatasia perinatal.
En algunos pacientes con diagnóstico reciente una transición abrupta de relativamente de apariencia normal diáfisis de metáfisis no calcificado aparece, lo que sugiere la aparición de cambios metabólicos bruscos.
Además, los estudios de serie de la radiografía puede mostrar la persistencia de la mineralización ósea disminuida (es decir, el raquitismo) y revelan la desmineralización generalizada gradual.
La mortalidad se estima en un 50% en el primer año de vida.
En la infancia hipofosfatasia expresión 's clínica es extremadamente variable. Como resultado de aplasia, hipoplasia o displasia de cemento dental, pérdida prematura de dientes temporales (es decir, antes de la edad de 5 años) se produce.
Con frecuencia, los incisivos se desprenden primera vez en casi toda la dentición primaria es exfoliada antes de tiempo. Las radiografías dentales a veces muestran las cámaras de pulpa agrandadas y canales de la raíz característica de los "dientes de cáscara" del raquitismo.
Los pacientes también pueden experimentar retraso en caminar, un andar característico de pato, se quejan de la rigidez y el dolor, y tiene una debilidad muscular apendicular (especialmente en los muslos) no progresiva en consonancia con miopatía .
Por lo general, las radiografías muestran deformidades raquíticas y característicos defectos óseos cerca de los extremos de los principales huesos largos (es decir, "lenguas" de radiolucidez que sobresale de la placa de crecimiento raquítico en el metaphsysis).
Retraso del crecimiento, fracturas frecuentes y osteopenia son comunes.
En los bebés afectados y los niños pequeños no es raro que, a pesar de la aparición de muy "abierto" fontanelas en los estudios radiográficos, para la sinostosis funcional de las suturas craneales que se produzca.
La ilusión de "abrir" los resultados de las fontanelas amplias zonas de la bóveda craneal hipomineralizada. Posteriormente verdadera fusión prematura de las suturas craneales óseas pueden elevar la presión intracraneal.
En edad adulta, hipofosfatasia puede presentar durante la mediana edad. Con frecuencia, hay antecedentes de raquitismo, la pérdida prematura de dientes temporales, o la pérdida temprana de la dentición adulta seguida de una salud relativamente buena.
Osteomalacia manifiesta en los pies dolorosos recurrentes derivados de mala cicatrización de las fracturas de metatarso el estrés y las molestias en los muslos o caderas debido a seudofracturas femoral que, cuando aparecen en el estudio radiográfico, se distinguen de la mayoría de los otros tipos de osteomalacia (que se producen en sentido medial) por su ubicación en la corteza lateral del fémur proximal.
Algunos pacientes sufren de calcio dihidrato declaraciones pirofosfato de cristal con ocasionales ataques abiertos de la artritis (pseudogota), que parece ser el resultado de una elevada endógenas pirofosfato inorgánico (PPi) los niveles.
Estos pacientes también pueden sufrir articular cartílago degeneración y la artropatía pirofosfato. Las radiografías pueden revelar seudofracturas en la corteza lateral del fémur proximal, fracturas por estrés, y los pacientes pueden experimentar osteopenia, condrocalcinosis, las características de la artropatía pirofosfato, y periartritis calcificada.
Odontohypophosphatasia está presente cuando la enfermedad dental es la única anormalidad clínica y los estudios radiográficos y / o histológicos no muestran evidencia de raquitismo o la osteomalacia.
A pesar de anomalías hereditarias de leucocitos y otros trastornos normalmente cuenta para esta condición, odontohypophosphatasia puede explicar algunas "de inicio temprano periodontitis "de los casos.
Perinatal e infantil hipofosfatasia se heredan como rasgos autosómicos recesivos con homocigosis o heterocigosis compuesta por dos alelos TNSALP defectuoso.
El modo de herencia de formas infantiles, adultos, y odonto de hipofosfatemia puede ser autosómica dominante o recesiva. Cuentas de transmisión autosómica por el hecho de que la enfermedad afecta a hombres y mujeres con igual frecuencia.
El asesoramiento genético es complicado por el patrón de la enfermedad de herencia variable, y por la penetración incompleta de la característica.
HPP es una enfermedad rara que se ha reportado en todo el mundo y parece afectar a personas de todas las etnias.
La prevalencia de hipofosfatemia severa se estima en 1:100.000 en una población de origen sajón Anglo en gran medida.
La frecuencia de HPP leve es más difícil de evaluar debido a los síntomas pueden pasar inadvertidos o ser mal diagnosticados.
La mayor incidencia de HPP ha sido reportada en la población menonita de Manitoba, Canadá, donde uno de cada 25 personas se consideran portadores y uno de cada 2.500 recién nacidos se manifiesta una enfermedad grave.
HPP se considera particularmente rara en las personas de ascendencia africana en los EE.UU.
TRATAMIENTO.
OSTEOSARCOMA
La denominación “osteosarcoma” se aplica a un grupo heterogéneo de neoplasias malignas de células fusiformes que tienen como rasgo común la producción de hueso inmaduro, también denominado “osteoide”. El grado de malignidad, y la consiguiente tendencia a metastatizar (o diseminarse), viene determinada por el grado histológico (esto es, la imagen que ofrece en el estudio microscópico). Esta familia de sarcomas incluye desde variantes en los que la curación queda garantizada únicamente con cirugía hasta casos letales, incluso tras las más agresivas medidas terapéuticas. A pesar de que las tasas de curación pueden aproximarse al 65-70% en casos de enfermedad localizada tratados con terapia multimodal, el tratamiento suele ser largo y arduo, y con frecuencia tiene una duración de un año o incluso superior. Dada la progresiva mejora en la tasa de supervivencia, de manera continua surgen nuevos nuevos desafíos en relación a los cuidados a largo plazo de los pacientes con osteosarcoma. Por consiguiente, el cuidado de los pacientes con osteosarcoma se lleva a cabo de forma óptima en un centro oncológico multidisciplinar, donde se dispone, de manera más directa y rápida, de los recursos y el personal requerido para el cuidado de estos complejos casos. El texto que sigue a continuación se centrará en el osteosarcoma de alto grado clásico, si bien, según proceda, se hará referencia a los subtipos existentes. Es también muy importante señalar que este artículo no pretende en ningún caso ser exhaustivo, sino ofrecer un resumen de los conocimientos actuales sobre el tema que faciliten la comunicación entre el enfermo y el médico.
En su artículo Osteosarcoma en la web eMedicine, Drs. Mehlman annd Cripe declaran: “El osteosarcoma es una enfermedad conocida de antiguo pero aún no comprendida por completo. El término sarcoma fue introducido por el cirujano inglés John Abernathy en 1804 y deriva de raíces griegas que significan “excrecencia carnosa” (Peltier 1993). En 1805, el cirujano francés Alexis Boyer (cirujano personal de Napoleón) usó por vez primera el término osteosarcoma (Rutkow 1993, Peltier 1993). Boyer concluyó que el osteosarcoma era una entidad diferente de otras lesiones del hueso como los osteocondromas (exóstosis).
Peltier L. F., "Tumors of bone and soft tissues" en Orthopedics: A History and Iconography. San Francisco, California, Norman Publishing; 1993: 264-291.
Rutkow I. M., "The nineteenth century" en Surgery: An Illustrated History. St. Louis: Mosby; 1993: 321-504.
Peltier L. F., "Tumors of bone and soft tissues" en Orthopedics: A History and Iconography. San Francisco, California, Norman Publishing; 1993: 264-291.
Rutkow I. M., "The nineteenth century" en Surgery: An Illustrated History. St. Louis: Mosby; 1993: 321-504.
Demografía del osteosarcoma
El osteosarcoma es el tumor primario óseo sólido más frecuente, constituyendo aproximadamente un 20% de los sarcomas primarios de hueso (Dahlin 1986). Es con mucho una enfermedad de la edad juvenil; más del 75% de los casos aparecen en pacientes menores de 25 años (Mirra 1989). Los casos aparecidos en adultos corresponden a sarcomas secundarios, esto es, sarcomas surgidos como complicación de enfermedades óseas preexistentes (enfermedad de Paget, osteomielitis crónica, infartos óseos) o sobre tejidos previamente irradiados. El osteosarcoma es algo más frecuente en varones, quizá debido a la mayor duración en ellos de la fase de crecimiento del esqueleto comparada con la de las mujeres (Dorfman 1988). La excepción a esta tendencia la constituye el sarcoma parostal, más frecuente en mujeres y en un grupo de edad ligeramente superior (Dahlin 1986). No se ha observado predilección por etnia o raza (Buckley 1998, Dorfman 1998, Weiss 1998).
Patogénesis y aspectos moleculares del osteosarcoma
¿Qué causa la aparición de un osteosarcoma? Aunque se ha conseguido cierto grado de conocimiento sobre la cuestión, la respuesta a esta pregunta continúa siendo un misterio en la mayoría de los casos. Fuchs y Pritchard (2002) dividieron los agentes causales “conocidos” en químicos, víricos, físicos (radiaciones) y otros (misceláneos). Los agentes químicos, que parecen actuar produciendo alteraciones genéticas, incluyen compuestos de berilio y metilcolantreno. Rous et al (1912) fueron los primeros en publicar la existencia de una causa vírica demostrable para los sarcomas. El virus del sarcoma de Rous (un retrovirus, esto es, un virus dotado de RNA) contiene un gen denominado V-Src, que tiene en las células normales un homólogo no patógeno, es decir, un proto-oncogén (Pritchard 1975). Así como algunos virus se han asociado con la inducción de tumores óseos, FJB es el único agente viral aislado de un sarcoma aparecido de novo (Fuchs 2002) y es conocido como un potente inductor de la aparición de osteosarcomas en ratones (Finkel 1966). El oncogén del FBJ está relacionado con un proto-oncogén (presente en condiciones normales en las células) llamado c-Fos (Fuchs 2002), que ha demostrado estar asociado a una pobre respuesta a la quimioterapia en pacientes con osteosarcoma (Kakar 2000).
La radiación parece desempeñar un papel crítico en la aparición de muchas neoplasias. Su papel en el desarrollo del osteosarcoma queda bien definida por su asociación con la aparición de sarcomas secundarios años después del tratamiento radioterápico de otras neoplasias, entre las cuales el osteosarcoma es una variante histológica habitual (Enzinger 1995, Tucker 1990, 1987, 1985, Huvos 1985, Weatherby 1981).
Se han sugerido otras causas de diversa índole. La asociación del osteosarcoma con la enfermedad de Paget del hueso es bien conocida, sucediendo en aproximadamente el 1% de los pacientes afectados por dicha enfermedad. Aunque el mecanismo exacto es aún desconocido, se ha sugerido como posible desencadenante una pérdida de heterocigosidad que afectaría al cromosoma 18 (Hansen 1999, McNairn 2001).
Una de las alteraciones genéticas asociadas con el osteosarcoma mejor caracterizadas es la pérdida de heterocigosidad del gen del retinoblastoma (RB). El producto de este gen es una proteína que actúa suprimiendo el crecimiento de las células con DNA dañado (esto es, se trata de un gen supresor tumoral). La pérdida de función de este gen permitiría a las células crecer de forma descontrolada, llevando a la aparición de diversos tumores, incluido el osteosarcoma. La presencia de esta mutación se ha asociado con tasas de supervivencia disminuidas en pacientes con osteosarcoma (Feugeas 1996). TGF-β es un factor de crecimiento cuyas cifras se elevan más en los osteosarcomas de alto grado que en las lesiones de bajo grado (Franchi 1998); es un bien conocido inhibidor del producto del gen RB contribuyendo quizá al comportamiento agresivo de estos tumores. También asociadas con el osteosarcoma están las mutaciones de p53, otro gen supresor; en la mayoría de los osteosarcomas se encuentra algún tipo de inactivación combinada de RB y p53 (Ladanyi 2003).
El receptor para el factor de crecimiento epidérmico (HER-2 o ERB-2) es otra alteración molecular asociada con el osteosarcoma. Su sobreexpresión se relaciona con tumores de curso clínico más agresivo, de potencial metastásico incrementado, intervalos libres de enfermedad más cortos y peores tasas de supervivencia global (Ferrari 2004, Morris 2001, Gorlick 1999, Onda 1996). Se han referido asociaciones similares para la glucoproteína P, un importante mediador de resistencia multidroga en las células tumorales (Ferrari 2004, Pakos 2003, Park 2001, Hornicek 2000), y para VEGF, un factor de crecimiento responsable de la angiogénesis tumoral (Hoang 2004, Kaya 2002, Zhao 2001, Kaya 2000). Si bien existen numerosas variaciones citogenéticas en los osteosarcomas, la presencia de patrones diagnósticos predecibles permanece ausente de forma generalizada (Sandberg 1994).
El receptor para el factor de crecimiento epidérmico (HER-2 o ERB-2) es otra alteración molecular asociada con el osteosarcoma. Su sobreexpresión se relaciona con tumores de curso clínico más agresivo, de potencial metastásico incrementado, intervalos libres de enfermedad más cortos y peores tasas de supervivencia global (Ferrari 2004, Morris 2001, Gorlick 1999, Onda 1996). Se han referido asociaciones similares para la glucoproteína P, un importante mediador de resistencia multidroga en las células tumorales (Ferrari 2004, Pakos 2003, Park 2001, Hornicek 2000), y para VEGF, un factor de crecimiento responsable de la angiogénesis tumoral (Hoang 2004, Kaya 2002, Zhao 2001, Kaya 2000). Si bien existen numerosas variaciones citogenéticas en los osteosarcomas, la presencia de patrones diagnósticos predecibles permanece ausente de forma generalizada (Sandberg 1994).
Presentación clínica del osteosarcoma
Los síntomas que con más frecuencia llevan a los pacientes con osteosarcoma a solicitar atención médica son el dolor y la aparición de una masa palpable (ver figura 1), evidenciable en hasta 1/3 de los pacientes en la primera visita (Widhe 2000).
En niños más pequeños la cojera puede ser el único síntoma. El dolor puede llevar muchos meses presente y ser inicialmente confundido con causas más corrientes como contracturas musculares, daño por sobrecarga o “dolores de crecimiento”. Con frecuencia no es hasta que se produce un traumatismo en la extremidad afecta y se realiza un estudio radiográfico cuando se evidencia la anomalía ósea. Desafortunadamente, si la fractura tiene lugar sobre el hueso debilitado por la neoplasia (la denominada “fractura patológica”), la tasa de recidiva local tras la cirugía se incrementa, y disminuye la expectativa global de supervivencia del paciente (Scully 2002). Un alto índice de sospecha acompañado de un cuidadoso examen de la articulación pueden disminuir la frecuencia de estos retrasos diagnósticos y los riesgos asociados.
El dolor que no se resuelve con medidas convencionales, su persistencia en reposo o el que despierta al paciente por la noche debe alertar al médico de la necesidad de realizar una evaluación más profunda. Una vez que se sospeche de la presencia de un tumor, la derivación del paciente a un oncólogo especialista músculo-esquelético debe de quedar garantizada.
Como sucede con la mayoría de los sarcomas, los pacientes habitualmente no se sienten ni parecen “enfermos”. La aparición de fiebre, mal estado general u otros síntomas constitucionales no son típicos del osteosarcoma. Los estudios de laboratorio pueden ser de ayuda, pero los resultados no son específicos del osteosarcoma. La tasa de sedimentación, los niveles de proteína C reactiva, fosfatasa alcalina (ALP) y lactato deshidrogenasa (LDH) pueden estar elevados. Se ha sugerido que la elevación pretratamiento de ALP, presente en aproximadamente el 50% de los pacientes, pudiera asociarse con un riesgo aumentado de recidiva (Bacci 1993). La elevación de la LDH se relaciona con un peor pronóstico, pues presumiblemente es indicativa de tumor con mayor agresividad biológica (Bacci 1994, Meyers 1992).
Características radiológicas del osteosarcoma
En la mayoría de las variantes de osteosarcoma una radiografía simple es prácticamente diagnóstica. Típicamente las lesiones se sitúan en las metáfisis (extremos) de los huesos largos, más frecuentemente en la rodilla (ver figura 2).
 |  |
Las lesiones son de límites mal definidos, acompañadas de desctrucción del hueso cortical y medular, y muestran osificación en el interior del componente de partes blandas (Gebhardt 2002, Gibbs 2001). Las lesiones pueden ser radiolúcidos, radiodensas o mixtas, dependiendo del grado de mineralización del osteoide (Kesselring 1982). Las variantes “de superficie” son diferentes, en tanto en cuanto parecen “descansar” sobre el hueso. La afectación destructiva del canal medular está ausente de forma típica en las lesiones “de superficie”, aunque pueden ser evidentes en enfermedad avanzada.
Los osteosarcomas telangiectáticos son con frecuencia completamente radiolúcidos, pudiendo ser confundidos con tumores líticos benignos como quistes óseos aneurismáticos. Si existiera cualquier duda, deberá realizarse una biopsia.
Otras modalidades de estudio de imagen tienen su papel en la evaluación inicial de una lesión sospechosa de osteosarcoma, especialmente la resonancia magnética nuclear (RMN). La RMN ha sustituido a la tomografía computerizada (TC) como prueba de elección en la determinación de la extensión de enfermedad local.
Aunque la TC detalla mejor la extensión de la destrucción ósea, la MRI tiene la ventaja de proporcionar imágenes axiales múltiples, un mayor detalle en la valoración del componente de partes blandas y en la relación del tumor con estructuras neurovasculares adyacentes. Es además más sensible a la hora de cuantificar la extensión de la afectación intramedular, ver figura 3 (Estrada 1995, Gillespy 1988, Sundaram 1987).
 |  |
Las proyecciones T1 coronal y sagital se utilizan para mostrar la extensión intramedular de la neoplasia, mientras que las proyecciones axiales en T2 permiten visualizar mejor el componente de partes blandas (Gillespy 1988). Además, la RMN con contraste permite una visualización precisa de la relación del tumor con las estructuras adyacentes (por ejemplo, nervios, vasos sanguíneos y músculos), convirtiéndola en una muy valiosa técnica para la estadificación y planificación quirúrgica. Como la RMN no conlleva la exposición del paciente a la radiación ionizante, proporciona además una vía segura y precisa para el seguimiento de la respuesta al tratamiento y el rastreo de recidivas mediante estudios periódicos (si bien las reconstrucciones con prótesis metálicas o placas óseas pueden afectar el detalle de imagen de la RMN).
La gammagrafía ósea (escintigrafía nuclear) y el FDG-PET son técnicas adicionales útiles, pero más adecuadas en la estadificación que en la evaluación de la lesión primaria. La mayor utilidad del escáner óseo en la evaluación del osteosarcoma radica en la detección de depósitos metastásicos en otros puntos del sistema esquelético.
Estadificación del osteosarcoma
Una vez sospechado un osteosarcoma, debe de realizarse una estadificación o estadiaje. Hay tres preguntas básicas que han de ser respondidas en esta fase:
- ¿Cómo es de agresivo el tumor (¿Cuál es su grado?)
- ¿Cuánto se extiende?
- ¿Se ha diseminado?
El grado hace referencia a la apariencia de agresividad biológica que muestra el tumor. Se basa en rasgos histológicos valorados en la toma biópsica. La mayor parte de los osteosarcomas son considerados de alto grado (de alta malignidad). La extensión se refiere a si el tumor ha crecido o no más allá de su compartimento de origen (en el caso del osteosarcoma, si ha erosionado y traspasado el hueso alcanzando las partes blandas adyacentes o no). La diseminación de cualquier tumor a otro lugar del organismo se denomina metástasis.
El estudio mediante RMN de la totalidad del hueso afecto es necesario no sólo para evaluar la extensión de la lesión primaria sino también para buscar metástasis ocultas (Van Trommel 1997), que pueden no ser vistas en el escáner óseo (Bhagia 1997). Estas últimas son focos metastásicos en el interior (o a distancia) del hueso de origen y aparecen en menos del 5% de los osteosarcomas (Campanacci 1999). Su presencia confiere un peor pronóstico pese a los continuos avances en la terapia adyuvante (Wuisman 1990, Sajadi 2004).
Los pacientes con enfermedad metastásica tienen generalmente un pronóstico peor que aquéllos sin metástasis detectables en el momento del diagnóstico. Es comúnmente aceptado que alrededor del 80% de los pacientes con osteosarcoma de alto grado tienen micrometástasis en el momento del diagnóstico, aunque no existen pruebas analíticas de sangre disponibles para la detección de este tipo de enfermedad metastática (Link 1986). En el estadiaje, el término metástasis hace referencia a aquéllas que pueden ser detectadas mediante técnicas de imagen (menos del 20% de los pacientes con osteosarcoma, ver Ferguson 2001). Los lugares más frecuentes de diseminación del osteosarcoma son el pulmón y el hueso. Por tanto, la radiografía y el TAC de tórax y el escáner óseo son esenciales en el proceso de estadificación.
Las técnicas “abiertas” hacen referencia a la cirugía, que se realiza en el quirófano. Estas proporcionan la mayor cantidad de tejido a evaluar por el patólogo, aunque frecuentemente no son necesarias, puesto que habitualmente puede realizarse una biopsia con aguja de la masa de partes blandas. Para que esta técnica sea útil es necesario contar con un patólogo experimentado en la evaluación de material obtenido mediante biopsia con aguja. Una vez elegida esta técnica diagnóstica, es el radiólogo el que, bajo control de imagen guiado con TC, la lleva a cabo. La colocación de la aguja puede ser dirigida por el cirujano, que además realizará la extirpación definitiva cirugía conservadora de la articulación (Mankin 1996, 1982).
Una vez realizada la evaluación patológica y asignado el grado histológico, toda la información es integrada a fin de definir la “personalidad” del tumor. Uno de los sistemas más habituales y sencillos utilizados en la oncología músculo-esquelética es la de Enneking et al (1986, 1980); ver Tabla 1.
| Estadio | Grado | Localización |
|---|---|---|
| IA | Bajo | Intracompartimental (en el compartimento óseo o muscular de origen) |
| IB | Bajo | Extracompartimental |
| IIA | Alto | Intracompartimental |
| IIB | Alto | Extracompartimental |
| III | Cualquiera + Metástasis | Cualquiera + Metástasis |
Utilizando este sistema, la mayor parte de los pacientes con osteosarcoma se encontrarían en un estadio IIB de la enfermedad. Este implica la existencia de un tumor de alto grado con extensión a partes blandas y sin metástasis detectables.
Histología del osteosarcoma
El hallazgo histológico definitorio del osteosarcoma es la presencia de células fusiformes osteoblásticas de malignidad evidente productoras de osteoide (Figura 4). No obstante, las variantes son comunes. En la actualidad, la Organización Mundial de la Salud (OMS) admite tres subtipos de osteosarcoma convencional; osteoblástico, condroblástico y fibroblástico (Raymond 2002). En algunos casos es posible dar diagnósticos erróneos de condrosarcoma o fibrohistiocitoma maligno. La presencia de hueso compacto con células estromales de apariencia maligna, independientemente de la asociación o no a una matriz condroide o fibrosa, establece el diagnóstico de osteosarcoma.
Características microscópicas del osteosarcoma
 |  |  |
El osteosarcoma osteoblástico está constituido en el examen microscópico por osteoblastos de aspecto maligno dispuestos sobre una matriz constituida mayoritariamente por hueso compacto. El osteosarcoma condroblástico consta de una matriz de apariencia cartilaginosa dotada de lagunas en las que se disponen células fusiformes malignas. La variante fibroblástica tiene la morfología de una neoplasia maligna de células fusiformes, en las que el único indicador de que se trata de un osteosarcoma es el escaso osteoide identificable. De forma habitual el tumor tiene una morfología mixta. Aunque el conocimiento de estas variantes puede ayudar al patólogo a considerar el diagnóstico de osteosarcoma cuando la histología no es clara, no hay datos que sustenten la existencia de diferencias en el comportamiento clínico o en el pronóstico basados en criterios microscópicos (Marina 2004).
Existen otros subtipos histológicos con relevancia clínica. El osteosarcoma parostal es una variante superficial de bajo grado. Microscópicamente está compuesto por un estroma fibroso de bajo grado, con menos atipia celular y mitosis que el osteosarcoma convencional (Okada 1994). Puede además mostrar una cubierta cartilaginosa similar a la del osteocondroma (Wold 1990). En raras ocasiones puede originarse en él un sarcoma de alto grado (Sheth 1996, Wold 1984). El osteosarcoma periostal es una neoplasia de superficie de grado intermedio. Se origina con más frecuencia en la diáfisis de los huesos largos y habitualmente muestra histología condroide. El osteosarcoma telangiectático puede asemejarse radigráfica e histológicamente a un quiste óseo aneurismático. La presencia de atipia celular y de producción de osteoide, aunque habitualmente escaso, distingue y caracteriza a esta variante de alta malignidad (Wold 1990).
Tratamiento del osteosarcoma
En los últimos treinta años hemos asistido a un enorme progreso en el tratamiento del osteosarcoma. El reconocimiento de la importancia de la terapia multimodal sumado a los avances en las técnicas de imagen son en gran medida responsables de ello. No sólo se han conseguido mejorías drásticas en la supervivencia, sino también en la habilidad para llevar a cabo de forma segura procedimientos de conservación de la extremidad en la mayoría de los pacientes con osteosarcoma.
El tratamiento estándar de los pacientes con osteosarcoma convencional incluye una combinación de quimioterapia y cirugía. Existe cierta controversia acerca de la duración de la quimioterapia y de la idoneidad de su aplicación, después (adyuvante) o antes de la cirugía. La segunda de ellas es denominada quimioterapia “neoadyuvante”. En lo que sí existe consenso general es en que tanto la cirugía como la quimioterapia por sí solas son insuficientes para el tratamiento del osteosarcoma clásico de alto grado.
En el caso de variantes de superficie de bajo grado, la cirugía por sí sola puede ser curativa. Aunque la quimioterapia puede estar indicada en lesiones de grado intermedio, no está claro que sea una indicación universal y su aplicación debe basarse en la valoración individual de cada caso.
Quimioterapia
El osteosarcoma debe de ser considerado una enfermedad sistémica. Se estima que aproximadamente el 80% de los pacientes tienen enfermedad micrometastásica en el momento del diagnóstico, a pesar de que sólo en un 10-20% de los casos ésta puede ser inicialmente detectada con técnicas de imagen convencionales (Ferguson 2001). En este hecho se sustenta la utilización de quimioterapia sistémica. Se ha demostrado que la quimioterapia, cuando se combina con la cirugía, produce un claro beneficio en los casos de osteosarcoma. Los mejores resultados en cuanto a supervivencia se evidencian en pacientes con enfermedad no metastásica.
Una vez que se ha producido diseminación “detectable”, el tratamiento se hace mucho más dificultoso y los resultados son menos predecibles, si bien con quimioterapia agresiva y cirugía aún es posible obtener periodos de supervivencia prolongada en cerca del 50% de los casos.
En un estudio randomizado, Link et al (1991) encontraron periodos de supervivencia libres de enfermedad en el 17% de los pacientes tratados sólo con observación tras la cirugía frente al 66% de los pacientes que recibieron quimioterapia. Las diferencias de supervivencia entre ambos regímenes se incrementan con el tiempo (Link 1993).
Esta modalidad terapéutica es, en la actualidad, la predominante en la mayoría de los centros. A pesar de que la administración de neoadyuvancia retrasa la cirugía aproximadamente 3 meses, tiene ciertas ventajas. Así, permite la valoración del grado de necrosis tumoral (muerte de las células neoplásicas) en el momento de la resección. Esto ofrece una valiosa información sobre el comportamiento de un tumor en particular.
En general, porcentajes de necrosis superiores al 90% son consideradas una buena respuesta a la quimioterapia. Menos claro está qué conducta adoptar ante porcentajes de necrosis inferiores al 90%, dado que no se ha demostrado que el cambio de régimen quimioterápico en pacientes con pobre respuesta al tratamiento mejore la supervivencia global (Ferguson 2001).
Tradicionalmente la quimioterapia se administraba tras la cirugía. Con el surgimiento de los procedimientos de conservación del miembro utilizando prótesis metálicas (cuya producción lleva semanas), algunos centros comenzaron a dar quimioterapia previa a la cirugía, en un esfuerzo por comenzar el tratamiento lo antes posible. Esto llevó a considerar la terapia neoadyuvante con cirugía posterior como sistema de tratamiento preferible (Rosen 1979).
Otra ventaja de la quimioterapia preoperatoria es que muchos tumores experimentarán una “consolidación” o “condensación” o incluso “encogerán”, haciendo más factible y segura la resección quirúrgica. La quimioterapia preoperatoria, aunque por las razones previamente citadas se considera de ayuda, no ha demostrado ser capaz de mejorar la supervivencia global libre de complicaciones en los pacientes con osteosarcoma (Goorin 2003), tal y como se creía inicialmente (Rosen 1979).
Los protocolos habituales para el osteosarcoma incluyen fármacos como la doxorrubicina, metotrexate en alta dosis, cis-platino e ifosfamida. Efectos secundarios como la toxicidad cardíaca o la supresión de la medula ósea pueden requerir el reajuste o incluso la suspensión del régimen suministrado.
El estudio previo a la terapia, que incluye un ecocardiograma y analíticas sanguíneas, es realizado de rutina como punto de partida para evaluar la función cardíaca y renal antes de iniciar un tratamiento potencialmente tóxico. Antes de iniciar la quimioterapia, debe abordarse con el oncólogo una exhaustiva discusión referida a los fármacos a emplear , el tiempo de administración y la toxicidad/efectos secundarios potenciales .
Cirugía
El osteosarcoma es una enfermedad quirúrgica. Una vez detectado, el tumor debe de ser extirpado si se busca garantizar la curación. Con mayor frecuencia, ésto se realiza tras un periodo de quimioterapia. El principal objetivo de la cirugía es extirpar el tumor de manera completa y segura. Históricamente, la mayoría de los pacientes eran sometidos a una amputación. En los últimos 30 años, los procedimientos conservadores de la extremidad se han convertido en el estandar, principalmente debido a los avances concomitantes en quimioterapia y en sofisticadas técnicas de imagen. Estos avances han hecho posible la preservación del miembro incluso tras una fractura patológica, que previamente era una indicación absoluta de amputación (Scully 2002). Las técnicas de preservación de la extremidad pueden ofrecer en la actualidad tasas de control local y supervivencia a largo plazo equivalentes a las de la amputación.
Cuando se selecciona un procedimiento quirúrgico para un paciente concreto, el objetivo oncológico de extirpar el tumor debe siempre tener prioridad sobre la conservación de la función.
Si el tumor puede extirparse de forma segura, conservando simultáneamente la extremidad viable, la técnica de conservación de la extremidad puede ser la apropiada. Si existe afectación de grandes vasos o nervios, o en caso de que la extirpación completa del tumor conlleve una pérdida significativa de función, la amputación puede ser una mejor elección. Otros factores como la edad del paciente, el nivel de funcionalidad deseado, preferencias estéticas y el pronóstico a largo plazo deben ser tenidas también en cuenta. Durante el proceso, un diálogo prolijo entre el paciente, los familiares de éste y el equipo médico-sanitario es de absoluta necesidad a la hora de elegir la vía quirúrgica a practicar.
La cirugía requiere gran cantidad de planificación y evaluación preoperatoria. El paciente es estadificado de nuevo (tal y como se describe anteriormente) antes de la cirugía a fin de determinar qué cambios han tenido lugar en respuesta a la terapia sistémica. Habitualmente se realizan radiografías convencionales y una RMN de la lesión primaria además de un escáner corporal completo y un TAC de tórax. Estos nuevos estudios dan con frecuencia la información óptima para la planificación quirúrgica, y ayudan además a detectar y/o evaluar la presencia de cualquier otra nueva lesión o la existencia de metástasis. Debe tomarse tan pronto como sea factible una decisión sobre la cirugía recomendable, especialmente si va a realizarse preservación de la extremidad, puesto que las técnicas de reconstrucción pueden tardar semanas en planificarse.
Los procedimientos quirúrgicos se incluyen en tres categorías básicas: amputación, preservación de la extremidad y plastia-rotación. La amputación incluye la extirpación de la articulación con un margen de seguridad entre el extremo de la porción del miembro conservado y el tumor (ver resección "amplia" o "radical" en la Tabla 2).
| Intralesional | Curetaje, extirpación tumoral parcial |
|---|---|
| Marginal | El margen es la zona reactiva limítrofe, puede quedar tumor residual |
| Amplia | Extripación del tumor y de un rodete de tejido sano circundante |
| Radical | Extirpación del compartimento completo, incluye amputación |
El pronóstico funcional tras la amputación es variable y depende de varios factores. En amputaciones de la extremidad superior, los resultados tienden a ser mejores en pacientes más jóvenes con la articulación del codo intacta. Las amputaciones de la extremidad inferior son más complejas. En un estudio se encontró que la percepción de los pacientes con amputación de la extremidad mostraban una fuerte correlación con la comodidad del muñón, el estado de la extremidad contralateral, el ajuste y función del dispositivo protésico, la percepción de cómo otros ven a los pacientes con amputaciones y de la capacidad para practicar ejercicio (Matsen 2000).
Aunque existen diferencias entre la amputación y la cirugía conservadora de la extremidad, el pronóstico a largo plazo relativo a la función y satisfacción del paciente parece ser similar (DiCaprio 2003, Refaat 2002, Nagarajan 2002).
Aunque inicialmente es más barata, a largo plazo la amputación puede ser más cara que la preservación del miembro con endoprótesis debido al coste de las prótesis y a la necesidad de su sustitución periódica (Grimer 1997). La mayor ventaja de la amputación es quizá que es una única intervención asociada con pocas complicaciones globales. Los amputados tienen además mayor libertad para la práctica continuada de deportes porque no deben preocuparse por complicaciones asociadas a las prótesis o injertos asociadas a preservación del miembro como aflojamiento o fractura.
La preservación de la extremidad consiste en extirpar el tumor con un ribete de tejido circundante sano preservando entre tanto el aporte vascular y nervioso al miembro (ver “resección amplia” en la tabla 2). Una vez extirpado el tumor, el defecto esquelético debe ser reconstruido. Algunos defectos pueden ser extensos, alcanzando tamaños de 15-20 cm y requiriendo reconstrucciones complejas (DiCaprio 2003). Como opciones se incluyen las prótesis metálicas, los aloinjertos de hueso (de cadáver), el hueso vascularizado obtenido del propio paciente o recolocación del hueso resecado tras esterilización en un autoclave. La elección debe de tener en consideración la localización y el tamaño del defecto, el pronóstico funcional que se espera y los deseos del paciente y/o de su familia. En la actualidad, las reconstrucciones endoprostéticas, con aloinjerto o combinadas (endoprótesis-aloinjerto) son las más frecuentemente realizadas.
La reconstrucción endoprostética ha alcanzado gran popularidad en la cirugía de preservación de la extremidad. Consiste en sustituir el hueso extirpado por un implante metálico (ver figura 5). Esto elimina la necesidad de tejido óseo para que el hueso “cicatrice”, que sí es necesaria con los injertos. Habitualmente puede realizarse una rehabilitación más agresiva de forma más temprana. Las complicaciones de este tipo de reconstrucción incluyen el aflojamiento de los componentes y las heridas.
 |
El aflojamiento aséptico sucede más en pacientes con menos de 20 años y sustituciones largas del fémur (Unwin 1996). La supervivencia global de estos implantes publicada es del 80% a los 5 años, 65% a los 10 años y del 50% a los 20 years (Damron 1997, Unwin 1996, Malawer 1995). Las tasas de infección en estas series alcanzan el 13%.
Incluso si se evitan complicaciones catastróficas, la necesidad potencial de múltiples revisiones o procedimientos de alargamiento pueden ser abrumadoras. Una publicación reciente sobre 25 pacientes con reconstrucción endoprotésica revela que 10 de ellos (40%) requirieron al menos una revisión y que el tiempo medio hasta la primera revisión fue de 4.9 años (Tunn 2004). Ruggieri et al (1993) refieren una tasa de complicaciones del 63% en procedimientos de preservación de la extremidad frente al 0% de la amputación y del 44% para la rotación-plastia. Es importante para el clinic y para el paciente comprender los riesgos inherentes y la realidad a largo plazo de sus elecciones.
Los injertos de cadáver tienen el beneficio del aporte de material biológico al hueso huésped. El factor limitante es el momento de implantación del mismo. La quimioterapia ha demostrado impedir la unión del aloinjerto al hueso huésped (Hazan 2001). Una extensa revision de más de 800 reconstrucciones con aloinjerto reveló que una supervivencia de éste superior a 3 años se asocia a una excelente supervivencia a largo plazo del mismo (Mankin 1996). Las mayores barreras para el éxito en los primeros 3 años fueron las infecciones (incidencia del 11%), fracturas (19%) y las dehiscencias o falta de unión (17%). Los aloinjertos osteoarticulares (aquéllos que sustituyen una superficie articular) afrontan el riesgo adicional del deterioro articular. En las series previamente citadas, un 16% de los pacientes con aloinjertos osteoarticulares en la rodilla y el 20% de los de la cadera requirieron sustitución completa de la articulación una media de 5 años después de la implantación. Aspectos adicionales del uso de aloinjertos voluminosos son la transmisión de enfermedades y la respuesta inmunológica al material extraño. Aunque el “rechazo” tal y como se conoce en otros órganos trasplantados no sucede, la reacción inmunológica puede impedir la cicatrización del injerto retrasando su integración. Las restricciones por tamaño pueden ser también limitantes, particularmente en pacientes más pequeños o jóvenes, ya que los donantes son habitualmente adultos.
La plastia-rotación es una técnica intermedia entre la amputación y la preservación del miembro usado más frecuentemente en osteosarcomas del fémur distal. En esencia es una amputación intercalada en la que se conservan las estructuras neurovasculares y la porción distal del miembro (pierna), reconectándolas a la porción proximal (fémur proximal y cadera) tras extirpar el tumor. El segmento distal se gira 180 grados para que la articulación del tobillo funcione como la articulación de una rodilla, convirtiendo una amputación sobre la rodilla en una amputación bajo la rodilla a fin de posibilitar al máximo el uso de una prótesis (ver figura 6).
 |
Se consiguen habitualmente resultados funcionales excelentes en lo que se refiere a flexión de la “rodilla”, deambulación protésica e incluso desarrollo de actividad deportiva (Merkel 1991). Esto es debido en parte a la conservación de la sensibilidad somática y propioceptiva del pie (Winkelmann 1996). La plastia-rotación se adapta especialmente a pacientes con esqueleto inmaduro (menores de doce años) con tumores adyacentes a la rodilla, aunque ha sido realizada con éxito en pacientes de más edad. La principal desventaja de esta forma de reconstrucción es la apariencia estética. La instrucción y el consejo preoperatorio sobre la naturaleza de la operación, el curso de la terapia física post-operatoria y la apariencia de la extremidad tras la plastia-rotación son esenciales. Con frecuencia es de gran ayuda que el paciente y su familia puedan conocer a alguien a quien se le haya realizado una plastia-rotación para obtener un mejor y más realista grado de entendimiento sobre cómo vivir con una extremidad rotada.
En pacientes con esqueleto inmaduro deben de considerarse ciertos aspectos particulares, puesto que el crecimiento continuado de la extremidad no afecta puede generar problemas adicionales y requerir múltiples operaciones ulteriores. Esto es especialmente exagerado en tumores originados en torno a la rodilla, ya que sus placas de crecimiento proporcionan el 70% de la longitud total de la extremidad. Puede incluso ser necesario el bloqueo de la placa de crecimiento contralateral o el alargamiento de la extremidad ipsilateral para equilibrar las diferencias de longitud de los miembros. Para conseguir la igualdad de longitud de la extremidades se están empleando, cada vez con más frecuencia, endoprótesis modulables y expandibles. Estos implantes utilizan ejes lisos no cementados que se fijan a través de la placa de crecimiento conservada a fin de permitir que continúe, al menos en parte, su desarrollo (Neel 2004, Eckardt 1993). Alcanzada la madurez esquelética, con frecuencia es necesaria la reevaluación para colocar una prótesis más “permanente”, con todos los riesgos asociados que conlleva, como se expuso previamente. Los riesgos son similares a los de otros procedimientos de conservación del miembro, siendo el aflojamiento y la infección los más frecuentes. A pesar de la necesidad potencial de realizar multiples procedimientos, los resultados funcionales y la satisfacción global de los pacientes parecen ser aceptables (Neel 2003, Plötz 2002, Eckardt 2000, Tillman 1997, Ward 1996, Eckardt 1993, Kenan 1991).
Pronóstico
Con los regímenes de tratamiento habitual, los pacientes con osteosarcoma sin metástasis detectables tienen tasas de supervivencia que se aproximan al 70%. Los factores que parecen tener un impacto pronóstico negativo son la localización (las axiales tienen peor comportamiento), un mayor tamaño tumoral, una pobre respuesta a la quimioterapia y la presencia de enfermedad metastásica (Bielack 2002). La más consistente y clínicamente relevante de éstas es la presencia de metástasis detectables (Bielack 2002, Marina 1993, Meyers 1993). Los pacientes que se presentan con lesiones pulmonares resecables tienen una supervivencia del 30-50% (Bacci 1997). Aquéllos con metástasis pulmonares irresecables, lesiones que no responden a la quimioterapia o con lesiones óseas múltiples tienen un comportamiento mucho peor, independientemente del tratamiento (Ferguson 2001, Bacci 1996, Meyers 1993).
Supervivencia
Una vez completado el tratamiento, se requiere un cuidadoso seguimiento para detectar signos de recidiva, metástasis y complicaciones relacionadas con el tratamiento. Esto incluye un cuidadoso examen físico, radiografías del lugar primario de origen, estudios de imagen periódicos del tórax, escáner óseo y pruebas de laboratorio. Esta evaluación es realizada de manera periódica frecuente en el post-operatorio inmediato y con mayores intervalos en el tiempo en tanto en cuanto el paciente permanezca libre de enfermedad. Si se detecta una recidiva, debe garantizarse la realización adicional de cirugía y quimioterapia. En ella son aplicables los mismos principios que en la enfermedad primaria, aunque las tasas de supervivencia a largo plazo son inferiores (Ferguson 2001, Link 1991). Algunos datos sugieren que los pacientes con recaída temprana (menos de una año después de concluido el tratamiento) tienen un pronóstico peor que aquéllos que recaen más tardíamente (Ferrari 1997).
Conclusión
Durante los últimos 30 años el osteosarcoma ha pasado de ser una enfermedad de carácter irremediablemente fatal a un proceso potencialmente curable. La mejoría en la supervivencia a largo plazo derivada de los avances en la terapia sistémica han llevado al surgimiento de nuevos desafíos en el cuidado de estos pacientes. Como los pacientes diagnosticados de osteosarcoma viven más, la function y los parámetros de medida de calidad de vida han ido haciéndose más importantes de forma creciente. Las decisiones relativas al tratamiento deben ahora tener en consideración complicaciones que pueden surgir mucho más tardíamente en la vida del paciente. Se confía en que una mayor comprensión de la etiología y de los mecanismos patogénicos que tienen lugar en el osteosarcoma llevarán a nuevas e innovadoras opciones de tratamiento. La investigación continuada y en cooperación de los frentes clínico y de laboratorio es necesaria para que estos futuros avances se materialicen.
Hipercalciuria idiopática en el niño
RESUMEN
Con el propósito de facilitar la orientación y el estudio de los pacientes con hematuria macroscópica recurrente o microscópica persistente, dolor abdominal recurrente, disuria, infección urinaria recurrente o litiasis nefrourológica en las que puede estar involucrada la hipercalciuria idiopática, se hace esta presentación de forma sencilla, para que pueda ser empleada por el médico de atención primaria. La hipercalciuria idiopática es tan frecuente que afecta aproximadamente al 10 % de la población y ante sus síntomas que pueden ser muy variados, hay que pensar en esta alteración metabólica hereditaria que es la causa más frecuente de litiasis renal cálcica. En ocasiones con una orientación adecuada y pocos o ningún medicamento se puede evitar que los pacientes desarrollen una litiasis nefrourológica o que la enfermedad litiásica progrese, y a la vez, impedir que estos pacientes, niños principalmente, sean sometidos a investigaciones cruentas.
Palabras clave: Hipercalciuria idiopática, litiasis nefrourológica, infección urinaria recurrente, hematuria.
CONCEPTO
Es una anormalidad metabólica hereditaria caracterizada por una excesiva excreción urinaria de calcio con niveles normales de calcio en sangre.1 La excreción urinaria de calcio declina con la edad de la lactancia a la adolescencia, después aumenta entre la tercera y sexta década de la vida y desciende nuevamente después de los 55-60 años en ambos sexos.2,3
MANIFESTACIONES CLÍNICAS
La hipercalciuria idiopática puede tener manifestaciones clínicas muy variadas, tales como episodios de hematuria macroscópica recurrente o microscópica persistente, disfunción urinaria, dolor abdominal o en el flanco, litiasis renal, infección del tracto urinario, leucocituria no infecciosa y disminución de la densidad ósea. Es la causa más frecuente de litiasis renal cálcica.1,4
INCIDENCIA
Se presenta en 5 a 10 % de la población5,6, pero se ha demostrado un incremento un algunos países llegando a 12% en los países occidentales.7 En Irán se ha reportado en 11,4 % de la población aparentemente normal y en 30 % de los que padecen infección urinaria recurrente.8
HERENCIA
Se ha planteado una herencia autosómica dominante porque en el 80 % de los pacientes con hipercalciuria hay un familiar con urolitiasis, pero se ha señalado también su alta frecuencia en las pequeñas islas, donde la consaguinidad es más frecuente.9 Aunque se han propuesto varios genes y alteraciones genéticas, la identificación precisa no se ha logrado aún.10
Cuando los varones de varias generaciones tienen hipercalciuria, litiasis urinaria y proteinuria (con o sin insuficiencia renal) se debe sospechar nefrolitiasis hipercalciúrica ligada al cromosoma X o enfermedad de Dent.11
DIAGNÓSTICO
Cuando por las manifestaciones clínicas se sospecha una hipercalciuria idiopática, siempre que sea posible, el diagnóstico debe hacerse en base a la excreción de calcio en orina de 24 h. En el adulto se considera anormal la excreción de más de 250 mg (6,25 mmol) de calcio en la mujer y de 300 mg (7,5 mmol en el hombre).12 Estos criterios con más de 50 años son los que se mantienen actualmente.10,13 En el niño mayor de 2 años, más de 4 mg (0,1 mmol) por kg de peso se considera hipercalciuria. En el menor de 2 años es muy dificil, prácticamente imposible, la colección de orina de 24 h, por lo que se hace necesario recurrir a la determinación de calcio/creatinina en orina, con el paciente en ayunas, en un corto período entre una y otra micción.11
La relación normal de calcio creatinina en orina en ayunas varía con la edad:
- De 0-6 meses: menos de 0,8 mg/mg,
- De 7-12 meses: menos de 0,6 mg/mg y
- Más de 2 años: menos de 0,2 mg/mg.
La determinación de la relación calcio/creatinina en una sola muestra de orina puede ser desorientadora. Una muestra de orina en ayunas (primera micción del día) y una muestra después de 2 a 4 h de la ingestión de alimentos que contengan leche es muy orientadora. La relación calcio/creatinina puede incrementarse un 40% o más de 0,28 después de la ingestión de alimentos. Si en la muestra en ayunas la relación calcio/creatinina es menor de 0,2 no es necesario hacer estudio en orina posprandial por considerarse normal por debajo de esta cifra.11
No debe determinarse calcio en orina con la finalidad de diagnosticar hipercalciuria idiopática en pacientes con infección urinaria porque las pielonefritis agudas aumentan la excreción urinaria de calcio.11
DIAGNÓSTICO DIFERENCIAL
Hay que tener en cuenta las causas secundarias de hipercalciuria que pueden verse en la hipercalcemia, hipofosfatemia, acidosis metabólica, hipomagnesemia, acidosis tubular renal, enfermedad de Dent y síndrome de Bartter.
Algunos medicamentos se han asociado con hipercalciuria como corticosteroides, furosemida, vitamina D y metilxantinas. No debe tratarse de diagnosticar hipercalciuria idiopática mientras el paciente esté recibiendo uno de estos medicamentos.11
EXÁMENES COMPLEMENTARIOS
En el párrafo sobre "diagnóstico", se explicó la determinación de calcio en orina para el diagnóstico de la entidad.
Para el diagnóstico diferencial es necesario realizar:
- Calcio y fósforo en sangre.
- Sodio, potasio, magnesio y gasometría en sangre.
- Sodio en orina. Si está elevado y se sospecha una alta ingestión de sodio, debe repetirse la prueba después de dos a cuatro semanas de restricción de sodio (2-3 g/día).
- Niveles de citrato en orina(valores normales: más de 40 mg/g de creatinina en niños de edad escolar).
- Ultrasonido renal.
- Creatinina.
- Ácido úrico (valor normal: menos de 0,56 mg/dL) y oxalato en orina (valor normal: menos de 50 mg/1,73 m2 de superficie corporal). Estas dos valoraciones solo se harán cuando haya criterio para su indicación.
- Densitometría osea (cuando esté indicada).
MANEJO DEL NIÑO CON HIPERCALCIURIA IDIOPÁTICA
- Explicación adecuada a los padres o tutores y al niño capaz de entender para que conozcan que es la causa más frecuente de litiasis nefrourológica.
- Aumentar la ingestión de líquido (40-45 mL/kg de peso por día).
- Reducir la ingestión de sodio.
- Introducir una dieta rica en potasio y baja en oxalato.
- Si con la restricción de sodio y el incremento de potasio, la hipercalciuria no se normaliza debe valorarse el uso de tiazidas, aunque sus efectos sobre los lípidos hacen cuestionable este tratamento por tiempo prolongado.
- Si existe hipocitraturia, agregar citrato de potasio al tratamiento.
- En ocasiones una orientación adecuada, con pocos o ningún medicamento, puede evitar la aparición de una nefrolitiasis o evitar la progresión de la enfermedad litiásica.
CONSIDERACIONES FINALES
La hematuria (macroscópica o microscópica) se presenta en más del 13 % de la población.14 La hipercalciuria es la causa más frecuente de hematuria asintomática en el niño,15 aunque en el adulto es menos frecuente y predominan las alteraciones del tracto urinario.16
Si se tiene en cuenta esta frecuencia y la conducta propuesta en busca de la hipercalciuria idiopática, se podrían evitar muchos procederes invasivos en estos niños.
LOS MARCADORES BIOQUÍMICOS ÓSEOS.
Los marcadores bioquímicos óseos son elementos extremadamente sensibles para el diagnóstico de perdedor y seguimiento terapéutico de la osteopenia, pero inespecíficos de enfermedad. Por lo tanto, todo hábito higiénico o dietético, medicamento o patología intercurrente que modifique el metabolismo óseo, afectará los valores de marcadores en el paciente.
En el momento de interpretar el resultado de los marcadores óseos, además de la densitometría ósea y la historia clínica del paciente debemos tener en cuenta cuatro puntos que sintetizamos en la tabla 1 .
Tabla 1 | |
Preparación del paciente | • para muestras urinarias reposo de ocho horas • para muestras sanguíneas, reposo de 8 horas, ayuno de 12 horas, extracción en las primeras horas de la mañana |
Oportunidad de toma de muestra | • el paciente no debe tener patologías virales o infecciosas intercurrentes • no debe presentar un accidente fracturario en agudo • no debe estar en reposo prolongado • no debe haber realizado ejercicio físico de alta intensidad el día previo |
Influencias constantes | • recordar que normalmente los valores de referencia dados son incompletos y no contemplan raza, etnia, edad, etc. |
Influencias variables | • medicamentos: diuréticos, corticoides, etc. • patologías primarias: mieloma, Paget, metástasis, enfermedades inflamatorias crónicas, endocrinopatías • cuadros intercurrentes: fracturas, virosis, artroplastias, infecciones, desnutrición, etc. |
_______________________________________________________________ (*) Patólogo Clínico, Jefe del Laboratorio Clínico de Instituto Nacional de Reumatología, ex Prof. Adj. de Patología Clínica, integrante del Grupo de Estudio de Osteoporosis de la Sociedad Uruguaya de Reumatología.
• Respuestas a las preguntas más frecuentes
• ¿Deben coincidir los valores de marcadores realizados en orina y en sangre?
NO, porque la muestra de orina es una cinética de varias horas del metabolismo óseo en el cual pudieron pasar diferentes cosas que influenciaron la muestra, mientras que la muestra sanguínea es una instantánea del metabolismo óseo. No obstante, en correctas condiciones de preparación del paciente, normalmente coinciden.
NO, porque cada técnica, ya sea que estudie la osteorresorción u osteoformación, tiene, desde el punto de vista analítico, mayor o menor sensibilidad o precisión e incluso un coeficiente de variación diferente (no confundir con sensibilidad o precisión respecto del valor diagnóstico)
• ¿Deben coincidir los valores de los marcadores óseos con los datos densitométricos?
NO, porque mientras que la densitometría, e incluso el standard de oro la histomorfometría con doble marcación , estudian algunos puntos del esqueleto, los marcadores son la resultante del metabolismo de todo el esqueleto.
• ¿Cómo se debe considerar el bajo valor de C telopéptidos encontrado en algunos pacientes tratados con antirresortivos?
Es un hallazgo relativamente habitual y se debe a la propia acción del antirresortivo.
Como ayuda para el clínico diremos que, en más del 90% de estos pacientes, al suspender el tratamiento, los marcadores vuelven a los límites de normalidad. En suma, es índice de efectividad terapéutica.
Una vez establecida internacionalmente la utilidad de los marcadores óseos, y que aprendamos a interpretar sus variables, queda por contestar otra pregunta frecuente:
• ¿Cada cuánto tiempo deben ser realizados los controles?
Para los marcadores de resorción consensualmente se establecen tres meses, y para los de formación seis meses. Una vez realizada la toma basal, se instaura el tratamiento y se realiza una nueva determinación entre 90 y 120 días. Si mejoraron los valores 30%, concluimos que la terapia es eficaz y tan sólo necesita un control anual. Si no hubo una buena respuesta: se revé al paciente y se realiza un nuevo estudio a los tres meses, una mejoría implica que la corrección terapéutica fue correcta y sólo debe controlarse al año.
• ¿Se obtienen mejores resultados usando más de un marcador, por ejemplo dos de resorción, o uno de formación y otro de resorción?
Como siempre pesa el costo-beneficio. Habitualmente obtenemos mejores resultados estudiando con profundidad pocos datos, que perdiéndonos en montañas de resultados paraclínicos.
Normalmente, basta con un marcador de resorción para el seguimiento, dado que el tratamiento está basado en antirresortivos, y dejamos, para casos particulares, los marcadores de formación. Por otra parte, estos últimos demoran normalmente seis meses en recuperarse, por lo cual pudimos haber perdido tiempo y dinero aplicando un tratamiento inadecuado, conclusión a la que se puede llegar más rápidamente con los marcadores de resorción.
• Influencia de medicamentos y cuadros intercurrentes sobre los marcadores
El objetivo de este capítulo es plantear nuevos conocimientos acerca de influencias variables o cuadros que puedan modificar el valor hallado de marcadores óseos, tanto en la muestra basal como en el seguimiento evolutivo y terapéutico.
Hoy sabemos qué debemos pensar ante una disociación de los resultados con la historia clínica, además de los ya mencionados y clásicos mielomas, Paget, etc., en:
• corticoterapia inhalatoria,
• artroplatias,
• virosis,
• anorexia / desnutrición.
Son temas nuevos, en desenvolvimiento, y pensamos que poco a poco, una vez que se conozca más detalladamente la riqueza y ductilidad del metabolismo óseo, se irán agregando más fenómenos que lo influencian en la lista.
• Corticoterapia inhalatoria
En materiales anteriormente publicados por la GEOSUR se trata la relación entre terapias corticoideas y osteoporosis. Se refiere principalmente a corticoides vía oral e intramuscular, definiendo o teorizando los mecanismos [2].
Debemos recordar que la absorción de corticoides es excelente también por piel y mucosas aunque, lamentablemente, cumplen con los mismos mecanismos que determinan sus efectos no deseados a nivel del metabolismo óseo.
Actualmente se usan corticoides inhalatorios como primera línea del tratamiento de cuadros asmatiformes crónicos, tanto en adultos como en niños. Cada vez se usan de forma más frecuente y a dosis mayores [3]. Es clara su ventaja dosis-beneficio respecto de los corticoides vía oral, en estos casos, sin embargo igualmente afectan el metabolismo óseo.
Si bien no se han observado complicaciones con dosis inferiores a los 1000 mcg/día, ya a estos niveles se encuentran elevaciones transitorias de marcadores de reabsorción , que pueden durar entre 24 a 72 horas. En tratamientos prolongados se ha descrito osteopenia, osteoporosis con fracturas a predominio axial y hueso trabecular.
Dada la frecuencia de estas terapéuticas y la dificultad de explicar al paciente que la inhalación no es inocua, por lo cual la dosis frecuentemente se autodetermina, en lo correspondiente a este capítulo diremos:
• en pacientes con terapia inhalatoria corticoidea, preguntar dosis, última inhalación y tiempo del tratamiento, dado que modifican rápidamente los marcadores bioquímicos óseos
• no deben realizarse determinaciones de marcadores óseos en las 24 a 48 horas posteriores a una corta terapia inhalatoria
• en la terapia crónica, debe realizarse la toma de muestra para marcadores óseos de forma lo más diferida posible de la última inhalación
• Efecto de las virosis sobre los marcadores metabólicos óseos
Ya se había observado que durante el curso de cuadros virales, el valor medio de los marcadores de remodelación ósea era comúnmente mayor. Este aumento se atribuía al reposo o menor movilización de la persona dado la astenia que habitualmente acompaña a éstos.
Actualmente se ha detectado la liberación de varias citoquinas proinflamatorias con acción inhibitoria de la osteogénesis y sobre todo estimulantes de la osteoclastolisis. Es así que se detectan:
• citoquinas proinflamatorias: aumento de los niveles de factor de necrosis tumoral alfa de hasta seis veces, de interleuquina IL 1 de hasta cinco veces, así como aumentos más o menos aumentados de IL6 e IL2,
• factor nuclear ligante kappa B,
• factor estimulante de colonias.
Vemos así que con un cuadro viral intercurrente NO deben estudiarse los marcadores bioquímicos óseos. El testeo de estos debe postergarse por un período no menor a quince días luego de la total recuperación del paciente. Si bien recordamos que aún no se ha determinado con exactitud: cuándo, cuánto, por qué período, ni cuál es claramente el mecanismo por el cual se altera el metabolismo óseo durante un cuadro viral intercurrente.
• Osteolisis periprotésica
En los pacientes con artroplastias, normalmente tienen valores de recambio óseo mayores que el adulto sano [4]. Eso se debe a:
• formación de citoquinas inflamatorias periprotésicas,
• estimulación de la osteoclatogénesis,
• osteolisis periprotésica.
¿Qué es lo que debemos esperar? El paciente con artroplastias puede tener valores basales de marcadores bioquímicos más elevados que el paciente osteoporótico sin prótesis y, sobre todo, no se puede pretender que una vez tratado vuelva a los niveles normales esperados.
Luego del tratamiento pueden mejorar hasta un 30%, pero habitualmente permanecen en cifras mayores a las esperadas para la categoría de rango de referencia del paciente sin prótesis.
• Anorexia y desnutrición
Tanto la anorexia, como la desnutrición crónica, tan poco tenida en cuenta durante la evaluación del paciente, se acompañan de un cuadro clínico marcado por: frecuente amenorrea, osteopenia, osteoporosis y elevación del recambio óseo [5].
Siendo que aproximadamente el 2%, entre niños y adolescentes, sufren de anorexia y que en este período se produce el mayor pico de masa ósea, (tabla 2) , que signará el futuro metabólico del paciente, este cuadro adquiere relevancia.
| Tabla 2 Formación de la masa ósea total | |
| Hasta los 3 años | 35% |
| Entre los 4 años y la pubertad | 20% |
| Adolescencia | 45% |
Es clara la repercusión de la anorexia y la desnutrición sobre el metabolismo óseo:
• grupos problema: 50 a 72% de los pacientes tienen densitometrías < - 2.5 desviaciones standard,
• grupos control: 22 al 28% de los pacientes tienen densitometrías entre -1 a 2.5 desviaciones standard,
• esto se acompaña de un marcado aumento del recambio óseo,
• esto se observa aun cuando la enfermedad dure un breve período de tiempo y la recuperación de los valores normales sea muy lenta.
La causa puede ser multivariable y, si bien se ha atribuido al déficit estrogénico, los mecanismos fisiopatológicos no están esclarecidos. No obstante, actualmente se toman las mismas consideraciones para la anorexia que para la desnutrición, pues durante ambas se observa:
• disminución de factor de crecimiento,
• marcada disminución del factor tipo insulino símil 1 (ILGF 1),
• lenta recuperación de los niveles de ILFG 1 que acompañan la recuperación del peso corporal y, a su vez, del metabolismo óseo.
Queremos destacar que la participación del ILGF 1 en la afectación del metabolismo óseo en estos casos estaría apoyada por una observación realizada en el año 1998 por un grupo de trabajo de científicos uruguayos; integrantes del Grupo de Estudio de Osteopatías de la Sociedad Uruguaya de Reumatología ( Dra. H. Havranek, Dra. G. Barreto, Dra. L. Labruna, Dr. Ramón Suárez ) en el Instituto Nacional de Reumatología; que pasamos a resumir:
Se tomó un grupo de 80 pacientes portadores de osteoporosis según los parámetros establecidos por la OMS, se les hizo un seguimiento clínico de 24 meses, en los que también se les realizó basal, a los 3 meses y cada año: OSTEOCALCINA, DESOXIPIRIDINOLINA, FOSFATASA ALCALINA ÓSEA, INTERLEUQUINA 1, FACTOR DE NECROSIS TUMORAL ALFA, ILGF 1 e ILGF 2. En todos los casos se realizó densitometría para ingresar al paciente a protocolo y fue repetida anualmente. En algunos casos se realizó histomorfometría con doble marcación, pre y postratamiento. Durante los 24 meses se administró una sustancia con probable efecto benéfico sobre la osteoporosis.
Los resultados obtenidos fueron:
• CLÍNICA: no hubo accidentes fracturarios durante los 24 meses,
• DENSITOMETRÍA: resultado indeterminado,
• HISTOMORFOMETRÍA: se comportó como antirresortivo,
• LABORATORIO: a) aumento paulatino de valores séricos de ILGF 1,
• mejoría de los marcadores de recambio óse.o
En suma: el ILGF 1 parece ser un elemento importante en la homeostasis del hueso. Esta relación directa observada en nuestro país, entre ILGF 1 y los marcadores bioquímicos, apoya la teoría de que durante la anorexia y la desnutrición la disminución de esta citoquina sea factor determinante para la instauración de la osteoporosis. Su significado adquiere importancia actualmente, cuando hay tratamientos específicos para corregir el déficit de ILGF.
En lo que nos ocupa en este capítulo diremos que ante un aumento inesperado de los marcadores óseos, tanto en la toma basal como durante el tratamiento o evolución del paciente osteoporótico, debemos pensar en su estado nutricional y despistar una posible anorexia .
3. Conclusión
Tanto durante la determinación basal como cuando se observan marcadas variaciones durante el seguimiento terapéutico con marcadores bioquímicos óseos, que no parecen coincidir con el cuadro clínico, debemos pensar: a) medicamentos que afectan el metabolismo óseo, b) patologías o cuadros clínicos intercurrentes.
Algunos de estos factores son siempre tomados en cuenta (mieloma, metástasis, fracturas, etc.). Mientras que de otros recién comenzamos a tener conocimiento: virosis, artroplastias, corticoterapia inhalatoria, anorexia, desnutrición, etc. Por lo tanto antes de pensar que es un error analítico, y reiterar la determinación, vía costo-beneficio, debemos preguntar por influencias variables.
Los marcadores bioquímicos óseos serán siempre una ayuda para el clínico cuando sean realizados en condiciones adecuadas e interpretados meticulosamente pensando que el paciente es un todo.
final de cuarta entrega....
quinta entrega.....
La absorción intestinal de calcio
1. Introducción
El calcio es un ion fundamental en los procesos de contracción muscular, secreción hormonal y permeabilidad de las membranas, razón por la cual el nivel sérico debe mantenerse en un estrecho rango fisiológico (8,5 - 10,5 mg/%).
El fósforo es fundamental en las uniones semiiónicas de alta energía, por lo que es el principal elemento de transmisión de energía intracelular (ATP), hace parte de la estructura de los fosfolípidos y ácidos nucleicos, y en el hueso participa en la fosforilación de la matriz proteica.
Desde el punto de vista evolutivo, cuando los seres biológicos se adaptaron a vivir fuera del mar, medio en el cual estos elementos eran abundantes, requirieron hormonas controladoras del calcio y desarrollaron la hormona parotiroidea, la hormona D (mal llamada vitamina D) y la calcitonina, encargadas de mantener los niveles de calcio, fósforo y magnesio en rangos fisiológicos.
Actualmente se conocen otros factores calciotrópicos como son el péptido relacionado con la paratohormona, las interleukinas 1, 2 y 6, el factor de crecimiento derivado de las plaquetas y la familia de factores de crecimiento, similares a la insulina y sus proteínas transportadoras.
2. Fisiología del calcio
El calcio constituye el 2% del cuerpo humano; sin tener en cuenta el hidrógeno, es el catión más abundante. El 99% de este elemento está en el esqueleto, en permanente equilibrio dinámico con el líquido extracelular, en el tracto digestivo y el riñón. El calcio es absorbido en el intestino delgado por difusión pasiva y transporte activo, controlado por la vitamina D. Aproximadamente se absorbe el 50% del contenido de calcio intraluminal, con una pérdida obligatoria por heces y por orina. Se filtra en un 100% y se reabsorbe en un 99% a nivel renal. El contenido total del calcio extracelular es de un gr, 40% unido a proteínas, especialmente albúmina, 47% iónico y el resto como bicarbonato y citrato.
Es necesario tener en cuenta la biodisponibilidad del calcio en los diferentes alimentos porque su contenido no siempre es utilizable. Está demostrado que el ácido oxálico forma complejos insolubles con el calcio que disminuyen su absorción, por ejemplo en la remolacha, el ruibarbo y el maní. De la misma manera el fitato, que se encuentra en las cáscaras de los cereales que tienen alto contenido de fibra, afecta la absorción del calcio por el mismo mecanismo.
Nutrición. La evidencia de la relación directa entre la dieta y la osteoporosis no está completamente definida. El pico de masa ósea parece depender predominantemente de factores genéticos; el mantenimiento del hueso sano, de factores nutricionales, y la pérdida ósea, de factores hormonales.
Calcio. El hueso es el depósito del 99% del calcio corporal, que allí tiene permanentemente actividades osteoblásticas y osteoclásticas, y cuyo resultado es un balance de cero en el adulto sano. Este balance del calcio dependerá del consumo y de factores absortivos con amplia variación individual; siempre existirá una pérdida obligatoria fecal, urinaria y por sudor.
Estudios del balance de calcio muestran que su absorción en niños y jóvenes es una función lineal del consumo hasta un nivel de saturación o umbral, arriba del cual, y el incremento del consumo no produce mayor acumulación ósea.
La absorción intestinal de calcio se realiza por dos mecanismos:
- Activo y saturable dependiente de la hormona D.
- Componente pasivo por difusión simple o facilitada mediante transportador.
En situaciones fisiológicas el intestino delgado es el encargado de absorber el calcio; esto se cumple de manera proporcional a la capacidad absortiva, la longitud del segmento intestinal, el tiempo de tránsito, la biodisponibilidad del calcio y la concentración de calcio intraluminal. El duodeno tiene la mayor capacidad absortiva, pero la mayoría del calcio se absorbe en el yeyuno.
A mayor cantidad de calcio en la dieta se absorbe más por el mecanismo de difusión, pero el transporte activo es saturable. En general se absorbe el 50% de la concentración intestinal de calcio intraluminal. Si se comparan las proporciones, a medida que aumenta el calcio en la dieta la proporción aumentará hasta un umbral, a partir del cual la fracción absorbida no se incrementa y empieza a disminuir (ver Tabla 1).
Tabla 1. Respuesta de adultos normales a varios grados de ingesta cálcica
Determinaciones Caso 1 Caso 2 Caso 3
Calcio de dieta (mg/d) 220 850 2.100
Calcio absorbido (mg/d) 150 340 490
Eficiencia (%) 68% 40% 23%
Excreción renal de calcio (mg/d) 150 210 260
Total balance de calcio (mg/d) -110 0 +70
Captación esquelética (mg/d) 420 420 420
Pérdida esquelética (mg/d) 530 420 350
Este mecanismo adaptativo de la absorción intestinal de calcio hace que la relación entre el calcio de la dieta y el calcio urinario no sea linear, y esto no se explica sólo por la saturación del transporte activo del calcio sino también por una respuesta inhibitoria de la hidroxilación de la vitamina D dependiente de PTH.
La absorción activa del calcio es regulada por 1-25 dihidroxi-vitamina d3, a su vez influenciada en forma positiva por PTH, HGH, estrógenos y progestágenos, y en forma negativa por glucocorticoides y hormona tiroidea.
El transporte activo del calcio disminuye con la edad más en mujeres que en hombres, debido a la deficiencia dietaria de vitamina D, a la mala exposición solar con disminución de la producción de ergosterol en la piel y a la disminución de la actividad enzimática de la hidroxilasa renal por envejecimiento y por deficiencia de estrógenos. Siempre existe pérdida obligada de calcio por riñón, heces y sudor, que en adultos sanos, sin alteración del recambio, es en promedio de 400 mg%.
Durante el crecimiento existe un aumento del calcio corporal del 0.2 al 2% y en contenido absoluto de 25 gr a 1.300 gr. El promedio de retención es de 40 mg/año hasta los 10 años; y hasta 400 mg/año son retenidos en la pubertad.
Fibra. El consumo de fibra se ha correlacionado en forma positiva con la densidad mineral ósea. Al parecer las dietas alcalinas tienen un efecto benéfico para el intercambio de calcio en el riñón, ya que la acidosis inhibe la reabsorción tubular del calcio.
Vitamina K. La deficiencia de vitamina K se ha asociado con la osteoporosis, pero esto no se ha aclarado; los niveles de vitamina K circulantes y de menaquinona son bajos en casos de fractura de cadera.
Las alteraciones de la carboxilación de la proteína Gla ósea (BGP), la hipercalciuria y el aumento de niveles de hidroxiprolina urinaria, en pacientes osteoporóticos, responden satisfactoriamente a la administración de dosis fisiológicas de vitamina K. La vitamina K es necesaria para la gamacarboxilación de los residuos del ácido glutámico en las proteínas, incluidas las que controlan la coagulación. Los residuos gamacarboxilados al parecer funcionan inhibiendo la mineralización y sirviendo como señales quimiotácticas osteoclásticas. Existen dos proteínas dependientes de la vitamina K: la osteocalcina y la nefrocalcina (proteína Gla renal).
Vitamina D. Es la vitamina más importante en el metabolismo óseo. Actúa como una hormona y ejerce las siguientes funciones:
- Facilita la mineralización ósea y la absorción intestinal de Ca y P.
- Regula la absorción renal y la homeostasis de aminoácidos y fosfatos.
- Influye en la actividad PTH de riñón y hueso.
- Estimula las actividades osteoblástica y osteoclástica en el hueso.
- Influye en la actividad del páncreas y en la función de la glándula tiroides.
- Modula la proliferación y diferenciación celular.
- Modula la función muscular.
- Es modulador del sistema paracrino y regulador de macrófagos; también de la secreción de citoquinas y la síntesis de inmunoglobulinas por linfocitos.
La deficiencia de la vitamina D induce la osteomalacia. En pacientes de edad, debido a subnutrición, o baja exposición solar y a disminución de la capacidad sintética de la piel para convertir el 7 dehidrocolesterol, se ha documentado déficit de vitamina D. El aporte de vitamina D facilita la absorción intestinal de calcio y fósforo, estimula la síntesis de osteocalcina por osteoblastos y promueve la diferenciación celular.
En la mayoría de los ancianos pueden ser beneficiosos 10 a 20 microgramos/día de vitamina D para la prevención de fracturas vertebrales y de cadera, aun en la ausencia de osteomalacia o deficiencia de vitamina D.
Otros. Estudios epidemiológicos han asociado la masa ósea a la ingesta dietética de elementos traza y vitaminas. Entre ellos se mencionan: zinc, vitamina B12, vitamina B6, silicón y magnesio. La vitamina C es cofactor del metabolismo normal del colágeno; el boro aumenta la fosfatasa alcalina. La disminución del manganeso produce deformidad ósea y alteraciones en la formación del cartílago, disminución del silicio conlleva alteración de la calcificación, y la disminución del vanadio produce deformación del esqueleto.
3. Rol del calcio en el pico de masa ósea
Con respecto al rol del calcio en la masa ósea, en el Consenso Mundial de Osteoporosis de 1993 se presentaron los estudios de los doctores Johnston y Lloyd, que mostraron aumento de la ganancia de masa ósea en niños suplementados, diferente de lo ocurrido a los no suplementados. En ambos estudios la ingesta de calcio fue mayor que la recomendada por la RDA norteamericana, con un ajuste de aproximadamente el 65%.
Sin embargo, aunque fueron estudios de tres años y se descartó la remodelación transitoria, no establecieron definitivamente si el mayor consumo de calcio conduce a llegar más rápido a determinado pico de masa ósea o simplemente lo aumenta. Heaney mostró un 13% de variabilidad en el contenido mineral óseo total de poblaciones genéticamente similares, lo cual sugiere un efecto ambiental superpuesto a la variabilidad genética. Cuando se administró calcio a gemelos monocigóticos se aumentó la densidad mineral ósea antes de la pubertad y la diferencia desapareció al suprimir el suplemento.
Es importante tener en cuenta que antes de la pubertad no existen diferencias significativas del requerimiento de calcio entre sexos. Respecto a la densidad mineral ósea del antebrazo, se empieza a notar diferencia entre hombres y mujeres en la edad adulta que se incrementa hasta un 30 a 50%. Ya hay suficiente evidencia para afirmar que una subnutrición en aquella etapa de crecimiento acelerado, especialmente con ingestas de calcio menores de 500 mg, modifica la expresión del pico máximo de masa ósea en modelo animales y humanos. El requerimiento entre los 9 y los 17 años es de 1.600 mg.
La relación entre menarquia y retención esquelética de calcio fue aclarada por los trabajos de Abrams, O'Brien y Stuff, en los cuales establecen que la máxima retención esquelética de calcio se presenta entre la pre y la postmenarquia, que disminuye durante el primer año de la menarquia y se estabiliza cuatro años después. La explicación de este fenómeno se halla en el efecto de los estrógenos sobre la mineralización de hueso nuevo y el subsecuente cierre epifisiario.
4. Rol del calcio en la menopausia
Un análisis de publicaciones entre 1966 y 1994, en búsqueda de la relación entre el consumo de calcio y la masa corporal, mostró un significativo coeficiente de correlación en mujeres premenopáusicas. Al suplementar con 1000 mg/día de calcio se previno en un 1% la pérdida ósea por año en todos los huesos, con excepción del cúbito.
Aunque en la menopausia temprana el déficit de calcio es dependiente del déficit estrogénico, el aumento de la remodelación aumenta las necesidades mínimas de calcio. Se observó que los niveles de osteocalcina eran modificados por el consumo de calcio, lo cual confirma que la dieta interviene en el recambio óseo.
La disminución de la relación calcio-proteína es un determinante negativo en la ganancia de masa ósea en mujeres desde los 30 años.
Ingesta proteica. La administración excesiva de proteínas aumenta el calcio urinario y produce un balance de calcio negativo, debido a la carga ácida resultante de los aminoácidos.
Existen diferencias individuales en el grado de pérdida ósea, en la velocidad de ésta y en el riesgo de fractura, pero se ha establecido un aumento en la velocidad de pérdida ósea en pacientes que muestran baja ingesta de calcio. El balance de calcio se considera como una función lineal de su consumo de calcio hasta encontrar un punto de equilibrio que estaría en 1000 mg en la premenopausia y 1.500 mg en la postmenopausia. Los trabajos del doctor Heaney muestran que entre 1.000 y 2.000 mg no hay mayor diferencia. Desde el punto de vista fisiológico está confirmado que dietas de menos de 400 mg de calcio en esta edad producen mayor pérdida de calcio.
Farmacos que alteran el estado nutricional
Los fármacos pueden afectar al estado nutricional del individuo a través de la alteración de los procesos de ingesta, absorción, metabolismo (afectando solo a las vitaminas) y excreción (hace sobre todo referencia a los minerales) de los nutrientes.
Pueden darse tres situaciones:
Pueden darse tres situaciones:
- Fármacos que provocan cambios en el apetito:
- Incrementando la ingesta.
- Disminuyendo la ingesta.
- Fármacos que alteran la percepción gustativa u olfativa.
- Fármacos que provocan náuseas y vómitos.
FÁRMACOS QUE INCREMENTAN LA INGESTA
Son generalmente fármacos utilizados en psiquiatría, que mejoran la función psicológica y conductual, provocando un aumento del apetito. Encontramos benzodiazepinas, fenotiazenas, antidepresivos triciclitos (amitriptilina) o terapias con antidepresivos triciclitos e IMAOs. Con estos tratamientos se pueden llegar a producir aumentos de peso importantes, no obstante en algunos pacientes provocan somnolencia, apatía y falta de interés por la comida.
También antihistamínicos H1, antiserotoninérgicos (astemizol, pitotifeno, ciproheptadina), hipoglucemiantes orales, anticonceptivos orales, estrógenos.
Por tanto el tratamiento con estos fármacos habría que vigilar el peso y la alimentación, debido a que pueden provocar obesidad al aumentar el apetito.
Son generalmente fármacos utilizados en psiquiatría, que mejoran la función psicológica y conductual, provocando un aumento del apetito. Encontramos benzodiazepinas, fenotiazenas, antidepresivos triciclitos (amitriptilina) o terapias con antidepresivos triciclitos e IMAOs. Con estos tratamientos se pueden llegar a producir aumentos de peso importantes, no obstante en algunos pacientes provocan somnolencia, apatía y falta de interés por la comida.
También antihistamínicos H1, antiserotoninérgicos (astemizol, pitotifeno, ciproheptadina), hipoglucemiantes orales, anticonceptivos orales, estrógenos.
Por tanto el tratamiento con estos fármacos habría que vigilar el peso y la alimentación, debido a que pueden provocar obesidad al aumentar el apetito.
FÁRMACOS QUE DISMINUYEN LA INGESTA
Son más problemáticos, siendo más difícil de evitar.
Son más problemáticos, siendo más difícil de evitar.
- Fármacos anorexígenos de acción central: son estimulantes del SNC. Se han utilizado en la terapia de la obesidad, pero no son recomendables por los efectos adversos, ya que pueden crear dependencia, taquicardia e insomnio. Encontramos anfetaminas, fenfluramina, teofilina, cafeína.
- Interferón α: se utiliza en niños de forma crónica.
- Fármacos anorexígenos de acción periférica: no actúan en el SNC, sino a nivel periférico, enlenteciendo el vaciado gástrico, aumentando la sensación de saciedad, dejando de ingerir alimentos.
Salbutanol, levo-dopa, antihistamínicos.
- Fármacos que producen anorexia por inducir un déficit nutricional: diuréticos, antiácidos (hidróxidos de Ca y Mg), fenitoina, sulfasalazina, estos últimos antifolatos.
- Fármacos que producen trastornos gastrointestinales (dolor calambres), provocan un malestar que disminuye el apetito. Aspirina, antiinflamatorios noesteroideos, suplementos de hierro (sobre todo los inorgánicos, al tolerarse mal, provocan molestias, disminuyendo la ingesta).
FÁRMACOS QUE ALTERAN LA PERCEPCIÓN GUSTATIVA U OLFATIVA
- Provocan la pérdida del gusto (ageusia).
- Disminuyen la sensación al gusto (hipogeusia).
- Provocan sensaciones gustativas anormales o desagradables (disgeusia).
- Provocan alucinaciones gustativas (pantogeusia).
- Provocan xerostomia (sequedad de boca): pueden recomendarse chicles o limón que alivian el problema.
Fármacos que alteran la percepción gustativa | |||
Ácido acetilsalicílico | Clofibrato | Insulina | Oxifedrina |
Ácido etacrínico | diazácido | Laurelsulfato sódico | Penicilamina |
Ampicilina | Dinitroenol | Licomicinas | Propanfelina |
Anfetaminas | Estreptomicina | Carbonato de litio | Tetraciclina |
Anfotericina B | Fenindiona | Maprobamato | Zidobudina |
Captoprilo | Fenitoina | Meticidina | Metromidazol |
Cloremiramina | |||
FÁRMACOS QUE PROVOCAN NÁUSEAS Y VÓMITOS
Muchos fármacos provocan nauseas o vómitos, en la mayoría de los casos transitorios, en otros casos es posible modificar el tratamiento. Estos casos no revisten problemática nutricional al revertir los efectos a los días.
Algunos fármacos provocan vómitos severos y permanentes, pudiendo afectar a la ingesta y la absorción de nutrientes como quimioterípicos citotóxicos en el tratamiento contra el cáncer: altretamina, carbopletino, carmustina, cisplatino, ciclosfamida, docarbacina, doctomicina, daunorubicina, doxorubicina, epirubicina, idorubicina, ifosfamida, irinotecam, lomustina, mecloretamina, fomustina, mecloretamina, mitoxantrona, pentostatina, esteptozocina.
También los antiretrovirales utilizados contra el sida provocan estos efectos, aunque no se sabe si es a causa de la enfermedad o por el tratamiento en si.
Muchos fármacos provocan nauseas o vómitos, en la mayoría de los casos transitorios, en otros casos es posible modificar el tratamiento. Estos casos no revisten problemática nutricional al revertir los efectos a los días.
Algunos fármacos provocan vómitos severos y permanentes, pudiendo afectar a la ingesta y la absorción de nutrientes como quimioterípicos citotóxicos en el tratamiento contra el cáncer: altretamina, carbopletino, carmustina, cisplatino, ciclosfamida, docarbacina, doctomicina, daunorubicina, doxorubicina, epirubicina, idorubicina, ifosfamida, irinotecam, lomustina, mecloretamina, fomustina, mecloretamina, mitoxantrona, pentostatina, esteptozocina.
También los antiretrovirales utilizados contra el sida provocan estos efectos, aunque no se sabe si es a causa de la enfermedad o por el tratamiento en si.
FÁRMACOS QUE ALTERAN LA FUNCIÓN GASTROINTESTINAL
- Fármacos que provocan esofagitis: doxiciclina, sulfato ferroso potasico, tetraciclina. Se puede evitar la esofagitis tomando los medicamentos con abundante agua o usando formas orales liquidas, disminuyendo el tiempo de contacto con el esófago.
- Fármacos que inducen pancreatitis: asparaginasa, azatioprina, didonosina, estrógenos, furosemida, diuréticos tiazídicos, valproato. Tras descartar que la pancreatitis no sea debida a otros factores (alcoholismo, cálculos biliares) debe sustituirse el tratamiento farmacológico.
- Fármacos que provocan colitis: antibióticos (al desequilibrar la flora bacteriana, se puede desplazar, aumentando la flora patógenas pudiendo dar lugar a colitis). Se trata con metronidazol o vancomicina.
- Fármacos que provocan diarrea: sorbitol (es vehiculo en algunas formulaciones por ejemplo líquidas de aminofilina, teofilina, acetoaminofeno, provocando la diarrea por un fenómeno osmótico). Antiácidos, laxantes. Se recomienda interrumpir el tratamiento o ajustar la dosificación.
FÁRMACOS QUE DISMINUYEN LA MOTILIDAD GASTROINTESTINAL
Alteran la absorción de nutrientes: puede ocurrir que se disminuya la motilidad, estando mas tiempo en el estomago degradándose a causa de los ácidos o que se vea aumentada la motilidad, disminuyendo el tiempo, disminuyendo por tanto su absorción.
Alteran la absorción de nutrientes: puede ocurrir que se disminuya la motilidad, estando mas tiempo en el estomago degradándose a causa de los ácidos o que se vea aumentada la motilidad, disminuyendo el tiempo, disminuyendo por tanto su absorción.
- Opiáceos
- Fármacos anticolinergicos.
Se recomienda la utilización de laxantes en caso de una intoxicación o la interrupción del tratamiento farmacológico en caso de que los laxantes no hagan efecto.
FÁRMACOS QUE AFECTAN A LA ABSORCION DE NUTRIENTES
- Fármacos eméticos.
- Fármacos que modifican el pH o las secreciones gastrointestinales.
- Fármacos que lesionan la mucosa intestinal.
- Fármacos que inducen hiperperistaltismo o constipación.
- Fármacos que inhiben específicamente la absorción de determinados nutrientes.
Fármaco | Mecanismo | Efecto |
Cimetidina | Inhibe la secreción acida gástrica | Malabsorción de vitamina B12 |
Antiácidos | Forman complejos insolubles | Malabsorción de fósforo |
Bicarbonato sódico | Alcalinizante | Malabsorcion de folatos |
Aceites minerales (laxantes) | Solubilización | Malabsorción de vitaminas liposolubles |
Laxantes en general | Alteran el transito gastrointestinal, alteran la mucosa | Malabsorción |
Colchicina | Daña la mucosa | Malabsorción |
Hipolipemiantes (colestiramina, colestipol) | Impiden la reabsorción de las sales biliares | Malabsorción de vitaminas liposolubles, B12, folatos, calcio, hierro y zinc |
RELEVANCIA CLÍNICA DE LAS INTERACCIONES QUE AFECTAN AL ESTADO NUTRICIONAL
La influencia del fármaco sobre el estado nutricional puede hacerse evidente desde el punto de visto clínico cuando:
La influencia del fármaco sobre el estado nutricional puede hacerse evidente desde el punto de visto clínico cuando:
- El fármaco se emplea de forma crónica, se mal utiliza o se abusa de él.
- El paciente presenta una limitación en los mecanismos de compensación (presenta una situación limite):
- Ingesta habitual insuficiente o marginal.
- Aumento en los requerimientos nutricionales debido a un estado catabólico.
- Absorción, metabolismo y excreción precarios o alterados por las patologías.
En muchos casos las manifestaciones clínicas de la interacción se atribuyen al proceso patológico primario (a la enfermedad en si), obviando y no evitando la posibilidad de una interacción (por eso estas interacciones al no estudiarse no se corrigen, en caso de hacerlo se podrían evitar y por tanto mejorar la situación).
canales cat1 cat2
Hasta hace poco tiempo, el endotelio que recubre internamente los vasos sanguíneos era visto como una capa de células que sólo cumplía labores de revestimiento. En los últimos veinte años, la investigación ha revelado que posee una extraordinaria variedad de funciones, sobre el control del tono arterial, la coagulación, la fibrinolisis, y el crecimiento vascular. Recientemente, la alteración de dichas funciones o "disfunción endotelial" ha sido implicada como un evento clave en la patogenia de la ateroesclerosis, la vasoconstricción coronaria y la isquemia miocárdica. La demostración que esta condición puede ser reversible abre la posibilidad de retardar la progresión de la aterosclerosis, reduciendo el riesgo de eventos cardiovasculares. Esto es importante dado la alta prevalencia de ateroesclerosis, que la convierte en principal causa de muerte en los países desarrollados.
A continuación presentamos 2 casos clínicos que ilustran sobre la etiología, el diagnóstico, el tratamiento y la presentación familiar de la disfunción endotelial.
Caso 1: Mujer 31 años, con antecedentes de pancreatectomía en 1985 por necrosis de un insulinoma no funcional. En 1992, a los 26 años, consulta por dolor precordial; su prueba de esfuerzo fue negativa. En 1993 cursa un embarazo con hipertensión arterial desde las 30 semanas, con un parto de término.
En 1996 consulta nuevamente por dolor precordial irradiado a brazo izquierdo y mandíbula. La coronariografía demuestra lecho coronario normal, con espasmo inducido por catéter en coronaria derecha. El estudio de lípidos mostró CT 295 mg/dl, C-HDL 34 mg/dl, TG 120 mg/dl, C-LDL 237 mg/dl. Se inicia tratamiento con lovastatina 20 mg/día, diltiazem 60 mg/día y aspirina 100 mg/día. En 1998, presentó una trombosis de la vena esplénica que motiva una esplenectomía. Se modifica tratamiento a diltiazem 90 mg/día, simvastatina 10 mg/día y aspirina 100 mg/día, con lo que remite la angina. El perfil lipídico muestra una mejoría significativa: CT 199 mg/dl, C-HDL 41 mg/dl, TG 64 mg/dl, C-LDL 145 mg/dl.
En enero 1999, se diagnostica un embarazo de 7 semanas y se suspende la simvastatina y diltiazem. En evaluación una semana más tarde, el Doppler de arterias uterinas mostró una resistencia vascular aumentada [sobre el percentil 95 para la edad gestacional]. La prueba de función endotelial mostró una dilatación reducida [3,0% al min; se considera normal > de 6%] (Figura 1, panel A). Se inicia tratamiento con L-arginina oral 6 g/día; una semana después presentó Doppler de arterias uterinas y umbilical normal, PFE con dilatación normal [16% al min] (Figura 1, panel B). En controles posteriores la paciente mantuvo Doppler de arterias uterinas dentro de rangos normales, y en las evaluaciones mensuales de PFE presentó dilataciones mayores a 8% al min. El embarazo evolucionó satisfactoriamente, resolviendo su parto a las 39 sem. a través de cesárea por una causa obstétrica, con un recién nacido sano de sexo femenino de 3450 g.
 | Figura 1. A. Imagen de la arteria braquial de paciente caso 1, muestra diámetro basal de arteria y su dilatación frente al estímulo de isquemia transitoria [3,0%], previo a intervención. B. Imagen de la arteria braquial paciente caso 1, muestra diámetro basal de arteria y su dilatación frente al estímulo de isquemia transitoria [16,0%] bajo suplementación con L-aginina. |
Caso 2: Niño de 5 años, asintomático, hijo de la paciente anterior, sin antecedentes mórbidos de importancia, producto de un embarazo de término que evolucionó con hipertensión arterial materna desde las 30 semanas; peso de nacimiento 4120 g. En agosto 1998, por el antecedente materno de hipercolesterolemia, se realiza estudio de lípidos que revela CT 353 mg/dl, C-HDL 55 mg/dl, C-LDL 252 mg/dl, TG 61 mg/dl. Se inicia tratamiento con dieta restringida en grasas totales, la cual es mal tolerada y provoca incremento ponderal insuficiente.
En enero 1999, se observa CT 306 mg/dl, C-HDL 40 mg/dl, C-LDL 246 mg/dl, TG 100 mg/dl. La PFE muestra dilatación de 4% al min (Figura 2, panel A). Se inicia tratamiento con colestiramina 4 g/día y restricción del consumo de grasas saturadas y colesterol, que es bien tolerado. La curva de crecimiento ha sido adecuada. En Mayo 1999: CT 239 mg/dl, C-HDL 48 mg/dl, C-LDL 179 mg/dl, TG 142 mg/dl. La PFE al mes de tratamiento muestra una dilatación de 12% al min. (Figura 2, panel B).
 | Figura 2. A. Imagen de la arteria braquial de paciente caso 2, muestra diámetro basal de arteria y su dilatación frente al estímulo de isquemia transitoria [4,0%], previo a intervención. B. Imagen de la arteria braquial de paciente caso 2, muestra diámetro basal de arteria y su dilatación frente al estímulo de isquemia transitoria [12,0%], bajo tratamiento hipolipemiante. |
Estos dos casos clínicos de hipercolesterolemia severa ilustran la presentación de la disfunción endotelial con manifestaciones trombóticas, espasmo coronario y preeclampsia en la madre y como hallazgo en su hijo.
Historia: Desde las primeras evaluaciones del rol del endotelio sobre la enfermedad cardiovascular, existían antecedentes del efecto vasodilatador producido por acetilcolina en anillos aórticos de conejo con endotelio indemne, mientras que en ausencia de endotelio se observaba un efecto constrictor. Además se había evidenciado que un gran número de vasodilatadores arteriales, incluyendo los agonistas colinérgicos muscarínicos como acetilcolina, ejercían su acción a través de la liberación de EDRF, posteriormente identificado como óxido nítrico (NO).
Ludmer y cols. en 1986, en arterias coronarias de sujetos portadores de ateroesclerosis demostraron la existencia de "vasoconstricción paradojal" secundaria a acetilcolina. Esta alteración sugiere por primera vez que "existe una disfunción endotelial, capaz de sobrepasar cualquier equilibrio entre sistemas constrictores y dilatadores que normalmente influyen sobre las arterias coronarias".
Actualmente se define la disfunción endotelial como la imposibilidad del vaso sanguíneo de aumentar su diámetro en respuesta a un estímulo conocido, ocasionado por una insuficiente generación de agentes vasodilatadores en el endotelio. En arterias sanas se observa dilatación dependiente del endotelio en respuesta a estímulos vasoactivos (isquemia, acetilcolina) mientras que en diferentes estados patológicos se ha demostrado ausencia de este efecto vasodilatador, o incluso un efecto constrictor, producto del predominio del efecto directo de acetilcolina sobre receptores muscarínicos del músculo liso subyacente (Tabla 1).
Fisiopatología: El endotelio es un tejido funcionalmente complejo, productor de múltiples agentes que actúan en forma endocrina, paracrina y autocrina que afectan la vasoregulación, proliferación de células del músculo liso vascular, agregación plaquetaria y adhesión de monocitos. La vasoregulación ocurre como resultado de un equilibrio entre la liberación de factores relajadores y constrictores. El factor relajador predominante es NO, sintetizado desde L-arginina a través del sistema de enzimas conocido como óxido nítrico sintasas (NOS) que convierte L-arginina y oxígeno en NO y L-citrulina (Figura). Se distinguen tres isoformas de NOS, dos de ellas constitutivas, que se expresan en diferentes tejidos en condiciones fisiológicas o patológicas y son dependientes de calcio, la endotelial (eNOS) y la neural (nNOS); la isoforma inducible (iNOS) está presente en macrófagos y células inflamatorias. Fisiológicamente, la isoforma inducible se expresa en muy bajas concentraciones y su transcripción puede ser inducida por citokinas como interleukina-1, factor de necrosis tumoral y factores de crecimiento1.
 |
| Figura 3. Esquema de la interrelación del endotelio con factores circulantes, hemodinámicos y el músculo liso subyacente. Abreviaturas: AA: ácido araquidónico; ADP: adenosín difosfato; AMPc: adenosín monofosfato cíclico; GMPc: guanilato monofosfato cíclico; CT: colesterol total; C-HDL: colesterol unido a lipoproteínas de alta densidad.; C-LDL: colesterol unido a proteínas de baja densidad; COX: ciclo-oxigenasa; EDRF: factor relajador del endotelio; EDHF: factor hiperpolarizante derivado del endotelio; IL: interleukina; NO: oxido nítrico; PGI2: prostaciclina; P450: citocromo P450; PFE: prueba de función endotelial; TG: triglicéridos; TNF: factor de necrosis tumoral |
Los estímulos que activan la eNOS incluyen trombina, ADP, bradicinina, sustancia P, agonistas muscarónicos, roce tangencial y corriente circular. El aumento de actividad de eNOS producto del roce tangencial de la sangre contra la capa endotelial (shear stress), contribuye a la vasodilatación mediada por flujo, un importante mecanismo de autorregulación por el cual aumenta el flujo sanguíneo en respuesta al ejercicio. Una vez producido, el NO difunde desde la célula endotelial al músculo liso subyacente, en el que activa el sistema enzimático de guanilato ciclasa soluble, que al unirse a NO aumenta la concentración de GMPc, responsable final de los efectos celulares inducidos por NO. El GMPc genera disminución en la concentración de calcio intracelular, a través de activación de proteínas kinasas sensibles a GMPc que fosforilan proteínas claves como canales iónicos, bombas y enzimas que llevan a la relajación del músculo liso subyacente (Figura 3). De este modo la vía NO-GMPc es la mediadora de los efectos relajadores de muchas hormonas, incluyendo histamina, nitro-vasodilatadores, acetilcolina, estrógenos, isoproterenol e insulina. Existen otros factores de relajación que incluyen a prostaciclina y al factor hiperpolarizante (cuya estructura es desconocida), que son antagonizados por factores constrictores también liberados por el endotelio tales como endotelina-1, tromboxano, y prostaglandina H2.
Además de su rol vasodilatador, el NO (entre otras funciones) es un inhibidor de crecimiento y migración celular, de la expresión de moléculas proinflamatorias y de adhesión. Por este motivo, el endotelio normal tiene funciones anti-trombóticas y anti-ateroescleróticas, inhibe la agregación plaquetaria, la adhesión de monocitos y la proliferación del músculo liso vascular, todos factores de importancia en el desarrollo de ateroesclerosis y disrupción de la placa aterosclerótica. Por el contrario, en el endotelio disfuncional se induce la síntesis de moléculas quimotácticas y de adhesin para monocitos y linfocitos T y de factores que inducen diferenciación de monocitos a macrófagos. El endotelio disfuncional promueve la agregación plaquetaria a través de una menor disponibilidad de óxido nítrico y favorece la trombosis a través de una alteración de la relación entre activador del plasminógeno versus su inhibidor endógeno. Estudios in vitro han evidenciado que las células endoteliales provenientes de un endotelio disfuncional, presentan un estado protrombótico caracterizado por un aumento de la actividad de factor tisular y disminución de la actividad de proteína C.
Modelos experimentales de ateroesclerosis han demostrado una relación inversa entre disponibilidad de óxido nítrico y aparición de la enfermedad. La disminución experimental de la disponibilidad de óxido nítrico a través de un inhibidor de NOS aumenta el desarrollo de ateroesclerosis, mientras que el aumento de la disponibilidad de L-arginina, disminuye su desarrollo. Esto sugiere un importante rol del endotelio en la patogenia de la ateroesclerosis.
El descubrimiento que el endotelio vascular libera óxido nítrico a partir de L-arginina, determinó una nueva era en el estudio de este aminoácido tradicionalmente considerado como precursor de síntesis proteica, urea y creatina. Actualmente se sabe que muchos tipos celulares utilizan L-arginina para liberar óxido nítrico. A pesar que las vías de síntesis y transporte de arginina son elementos clave en su metabolismo, las vías catabólicas han concitado el mayor interés ya que tres de sus productos finales son utilizados como moléculas señales: NO, glutamato y agmatina. Además, arginina por sí misma cumple roles como cofactor en el metabolismo de otros aminoácidos, siendo también capaz de regular la secreción de hormonas como insulina, glucagón, hormona de crecimiento y prolactina.
La mayoría de las células obtienen la arginina necesaria desde el medio extracelular a través de proteínas transportadoras. El mecanismo de captación preferente para arginina es el sistema y+, un transportador Na-independiente, de alta afinidad para arginina, lisina y ornitina. Recientemente se ha aislado el cDNA para dos proteínas transmembranas, CAT-1 y CAT-2, que exhiben propiedades de transporte de aminoácidos consistentes con el sistema y+. De ellas CAT-1 es constitutiva y exhibe una Km para arginina de 70 a 100 µM; es la más ampliamente distribuida, está presente en casi todos los sistemas excepto en hepatocitos. Sin embargo, el sistema CAT-2 (con peculiares características de Km y Vmax elevadas) puede ser inducido por citokinas inflamatorias en hepatocitos y otros tipos celulares; al mismo tiempo se induce una isoforma de sintetasa de óxido nítrico (iNOS), indicando que se incrementa el transporte de arginina para sustentar la producción aumentada de NO9,10.
Estudios recientes han mostrado que la localización celular de los transportadores de arginina es responsable de lo que se ha denominado la "paradoja de la arginina". Hace mucho tiempo se observó que la síntesis endotelial de NO puede ser regulada por concentraciones variables de arginina extracelular, a pesar que la concentración intracelular (0,1-1mM), excede en varios órdenes de magnitud la Km de la NOS endotelial para L-arginina (2,9 µM). La Km aparente de síntesis de NO por células intactas está entre 73-150 µM de L-arginina extracelular, lo que está dentro del rango de valores de Km para los transportadores de arginina (100-150 µM) y coincide con las concentraciones plasmáticas de arginina. Caveolina-1, proteína estructural de membrana plasmática y parte fundamental de invaginaciones o caveolas de membrana, tendría un rol en la regulación producida por cambios mecánicos (shear stress) sobre la actividad de eNOS y canales iónicos. Estudios inmunohistoquímicos han demostrado que CAT-1, eNOS y caveolina están co-localizados en cavidades de la membrana plasmática, sugiriendo una canalización preferente de arginina extracelular hacia eNOS. Esta peculiar dependencia de los niveles plasmáticos de arginina para la síntesis endotelial de NO, ha permitido que se utilice la suplementación de L-arginina como un método para favorecer la disponibilidad de NO.
CONCENTRACIONES VINCULADAS A DISFUNCIÓN ENDOTELIAL
1. Aterosclerosis y factores de riesgo cardiovascular. La enfermedad aterosclerótica es una de las principales causas de morbilidad y mortalidad en los países desarrollados y en nuestro país. En los últimos años, el conocimiento de su patogenia ha aumentado, en especial en lo que se refiere al rol del endotelio en la homeostasis vascular. Dada su localización, el endotelio es blanco primario de las fuerzas mecánicas y de las alteraciones humorales relacionadas con los factores de riesgo cardiovascular, como el tabaquismo activo y pasivo, diabetes mellitus, hipertensión arterial, dislipidemia e hiperhomocist(e)inemia. Estos factores de riesgo condicionan una alteración de las funciones normales del endotelio, favoreciendo la vasoconstricción, agregación plaquetaria y trombosis, aumento de la permeabilidad, adhesión y proliferación celular, condiciones que llevarán finalmente a la formación de la placa aterosclerótica y a su trombosis y/o ruptura.
Se ha demostrado que la disfunción endotelial, tanto en la arteria braquial como en las coronarias se asocia a la presencia de los factores de riesgo cardiovascular reconocidos, aun cuando no exista evidencia demostrable de ateroesclerosis. Esto sugiere que esta condición ocurre precozmente en el proceso de la enfermedad cardiovascular y que representa una anormalidad sistémica del endotelio. Más aún, en pacientes con enfermedad ateroesclerótica demostrada y/o disfunción endotelial, existe reducción de eventos clínicos adversos y reversión del proceso de disfunción del endotelio con intervenciones terapéuticas sobre diferentes factores de riesgo cardiovascular (Tabla 1). Los estudios clínicos han mostrado alteraciones en la respuesta del endotelio en arterias con enfermedad ateroesclerótica demostrada y en vasos sanos de pacientes que tenían ateroesclerosis en otra localización, lo que lleva a postular que la disfunción endotelial es una alteración sistémica que ocurre precozmente en el proceso de la enfermedad y es, por lo tanto, una vía común por la cual los factores de riesgo llevan al desarrollo de la enfermedad ateroesclerótica. En una segunda etapa, las placas ateroscleróticas condicionan una mayor disfunción del endotelio, propagando así el daño vascular.
| Condiciones asociadas a disfunción endotelial | Intervenciones que mejoran la función endotelial |
| Edad avanzada | Antioxidantes |
| Historia familiar de enfermedad cardiovascular | Antagonistas del calcio |
| Sexo masculino | Disminución del colesterol |
| Alimentación de alto contenido graso | Disminución de la homocisteína |
| Colesterol elevado | Embarazo fisiológico |
| Diabetes mellitus | Ejercicio físico |
| Hipertensión arterial | Estrógenos |
| Homocisteína elevada | Inhibidores de enzima convertidora de angiotensina |
| Obesidad | L-arginina |
| Tabaquismo | Suspensión del hábito de fumar |
| Deficiencia de estrógenos. | |
1.a Dieta: estudios realizados por Vogel y cols. evidenciaron que en sujetos sanos una comida hipergrasa, induce una alteración transitoria de la función vasodilatadora del endotelio, que se correlaciona con el incremento postprandial de lípidos plasmáticos, principalmente triglicéridos. En un estudio posterior, los autores demostraron que la suplementación previa de vitamina C y E previene esta alteración, sugiriendo un mecanismo oxidativo de daño endotelial. Un estudio realizado por Cuevas y cols., en 12 voluntarios sanos que recibieron durante 2 meses dietas mediterránea o una dieta rica en grasas saturadas y colesterol, observó que esta última se acompañaba de disfunción endotelial, que se corrigió con ingesta diaria de 200 ml de vino tinto por un mes. Ninguna de las dietas produjo modificaciones del peso corporal, presión arterial, o niveles séricos de glucosa y lípidos. El efecto favorable del vino, significativo sólo en condiciones de dieta hipergrasa, se atribuye al efecto antioxidante de flavonoides presentes en éste.
1.b Hipertensión arterial: En arterias aisladas de modelos experimentales de hipertensión se observa una reducción de la vasodilatación inducida por endotelio. El mecanismo que subyace a esta disfunción endotelial varía de un tipo de hipertensión a otro, y aunque se observa un deterioro de la vasodilatación inducida por endotelio en la mayoría de las formas de hipertensión, ésta no parece ser la causa de la elevación de la presión arterial. De hecho, la disfunción endotelial se observa en individuos normotensos provenientes de familias con alta incidencia de hipertensión esencial, por lo que podría ser un factor predisponente de hipertensión.
1.c Hiperhomocist(e)inemia: Homocisteína es un aminoácido sulfurado que se forma durante el metabolismo de metionina. El aumento de homocisteína plasmática se observa frecuentemente en ancianos, individuos con déficit de ácido fólico, cianocobalamina (vitamina B12) o piridoxal fosfato (vitamina B6), y en varias anormalidades enzimáticas incluyendo déficit de cistationin ß-sintetasa o metil tetrahidrofolato reductasa. En los últimos años se ha establecido que elevaciones de homocisteína plasmática producen un importante aumento del riesgo cardiovascular. En animales de experimentación la homocisteína reduce la biodisponibilidad de NO, lo que se ha visto corroborado con la demostración de falla en la vasodilatación mediada por flujo en la arteria braquial de sujetos hiperhomocisteinémicos y en sujetos sanos a los que se somete a una carga oral de metionina. En estos últimos el efecto se revierte con la administración de ácido fólico.
1.d Obesidad y resistencia a insulina: La insulina tiene una acción vasodilatadora específica en el músculo esquelético, la que parece tener importancia en la mantención del tono vascular y en la modulación de la absorción de sustratos. Se ha demostrado que el efecto vasodilatador está mediado por NO. En sujetos obesos que presentan resistencia a la insulina y en diabetes mellitus, la vasodilatación dependiente de endotelio se encuentra disminuida en 40-50% en relación con controles no-obesos. La respuesta vasodilatadora a la insulina también está deteriorada en estos pacientes.
2. Preeclampsia: en el embarazo fisiológico se observa una activación permanente de la función endotelial. Estudios iniciales muestran un aumento de la capacidad de dilatación en embarazadas respecto a controles no embarazadas. Por otro lado, la hipertensión arterial inducida por el embarazo o preeclampsia es la complicación médica más frecuente en el embarazo, afectando entre 5 y 10% de las mujeres. La patogenia de este desorden se relaciona con una anormalidad en la placentación, proceso a través del cual se forma una placenta funcional que permite el crecimiento y desarrollo del embarazo. Las primeras 20 semanas de gestación son claves en este proceso.
Para comprender cómo la disfunción endotelial puede ser causa de preeclampsia, y cómo la preeeclampsia puede causar o exacerbar una disfunción endotelial se hace necesario comprender la complejidad del proceso de placentación. Luego de la fecundación, el embrión ingresa a la cavidad uterina; en ese lugar el tejido trofoblástico que lo rodea se adhiere al epitelio endometrial. Posteriormente el trofoblasto procede a la penetración de la decidua basalis y del primer tercio de la pared muscular. En este proceso se genera una fijación mecánica al miometrio y se asegura un aporte de oxígeno y nutrientes al feto. El primer objetivo se logra con la penetración de células trofoblásticas al tejido conectivo uterino formándose las vellosidades de anclaje. Para cumplir el segundo objetivo, el tejido trofoblástico invasor guiado por un quimiotropismo positivo por el oxígeno se acerca a los vasos sanguíneos arteriales uterinos y penetra su pared en sentido lateral y frontal, llegando a la superficie interna del vaso donde reemplaza total o parcialmente la capa endotelial. La pérdida de la túnica muscular y elástica genera una reducción en la resistencia vascular. La consecuencia de este proceso es la generación de un espacio entre las vellosidades flotantes, denominado espacio intervelloso; en éste, la sangre de origen materno ingresa a una baja velocidad permitiendo que oxígeno y nutrientes difundan a la circulación fetal y retiren desde la circulación fetal al anhídrido carbónico y residuos del catabolismo fetal. Tanto el trofoblasto que invade las arterias maternas como el que tapiza las vellosidades flotantes se ha transformado en un equivalente del endotelio.
Este proceso, que requiere de una perfecta sincronización entre los distintos tipos celulares maternos y fetales, está alterado en la preeclampsia. Debido a causas aún desconocidas, en las que intervienen factores maternos, paternos y propios del embrión existe una menor capacidad invasora del trofoblasto la cual condiciona una reducida penetración y transformación vascular. Esto genera isquemia placentaria, proceso a partir del cual se producen diferentes condiciones que generan entre otros fenómenos, disfunción endotelial. La patogenia de la disfunción endotelial observada en la preeclampsia es múltiple y consecuencia de factores maternos o primarios y de origen placentarios o secundarios. Entre los factores maternos que se consideran "predisponentes" a la preeclampsia existen una serie de condiciones como trombofilia, hiperhomocisteinemia, diabetes mellitus, hipertensión arterial crónica, síndrome antifosfolípidos y Síndrome X. Estas condiciones maternas preexistentes, generan disfunción endotelial a través de los distintos mecanismos antes mencionados e interfieren en la interacción del endotelio materno con el trofoblasto fetal. Así, la disfunción endotelial basal se acentúa por factores placentarios como la liberación a la circulación de fragmentos de tejido trofoblástico necrótico que se adhieren al endotelio y de citokinas e intermediarios reactivos del oxígeno generados por la placenta isquémica.
Este análisis permite comprender la expresión clínica de la preeclampsia como reflejo de un defecto en las etapas iniciales del embarazo. Además, en el animal de experimentación, la reducción de la perfusión placentaria es capaz de provocar una elevación aguda de la presión arterial por mecanismos aún desconocidos, entre los que se postulan los factores placentarios que exacerban la disfunción endotelial. Este proceso es seguido de un aumento de la permeabilidad vascular sistémica y focal, en particular en el riñón, cerebro y la retina contribuyendo a las complicaciones de esta enfermedad. Por otra parte la menor perfusión placentaria es responsable de una restricción en el crecimiento fetal, y en casos más severos de una deficiente oxigenación al feto.
Hoy es posible evaluar en forma no invasiva la resistencia vascular uterina utilizando velocimetría Doppler en las arterias uterinas y la existencia de disfunción endotelial mediante ultrasonido vascular de alta resolución. En nuestra opinión, la importancia de esta aproximación reside en que la prevención y tratamiento de la preeclampsia debe considerar la intervención sobre la disfunción endotelial primaria o materna. El estudio previo a un embarazo planificado o en las etapas tempranas del embarazo debe realizarse en mujeres que han presentado preeclampsia severa, de inicio precoz, con importante restricción de crecimiento fetal o muerte fetal in utero.
El endotelio funcionante o disfuncionante ya sea de la vasculatura materna, de la placenta o de ambos territorios, se constituye en un elemento central que determina el curso fisiológico del embarazo o su desarrollo en condiciones isquémicas, con la expresión de preeclampsia, retardo de crecimiento intrauterino y parto prematuro.
3. Enfermedad cardiovascular en la vida adulta y enfermedad intrauterina: evidencias recientes demuestran correlación entre el peso de nacimiento y la dilatación endotelio dependiente en respuesta a la isquemia en arteria braquial. Al comparar en la vida adulta a pacientes que tuvieron un peso de nacimiento inferior a 2500 g. versuspacientes con peso de nacimiento normal (3000-3800 begin_of_the_skype_highlighting 3000-3800 end_of_the_skype_highlighting g), se observa que aquellos con bajo peso de nacimiento muestran una menor respuesta vasodilatadora, controlando otros factores como niveles de colesterol y hábito de fumar. Estudios de cohortes han demostrado asociación entre marcadores de bajo peso de nacimiento y una mayor mortalidad cardiovascular en el adulto. Además, el bajo peso de nacimiento y otras mediciones de crecimiento intrauterino se han relacionado con efectos desfavorables sobre la presión arterial, tolerancia a la glucosa, lípidos sanguíneos y factores de coagulación tanto en niños como en adultos. Estas observaciones han llevado a plantear que el riesgo cardiovascular del adulto tiene su origen en las primeras etapas de la vida, posiblemente en el período intrauterino.
Hallazgos recientes han sugerido que la adaptación fetal a una disponibilidad limitada de nutrientes determina cambios permanentes en su fisiología y metabolismo. Estos cambios "programados" podrían ser el origen de alteraciones permanentes en el desarrollo de los órganos, incluyendo estructura y función del aparato cardiovascular. Hasta ahora no está claro en qué consisten tales cambios y cuál es su curso temporal. El mecanismo fisiopatológico básico involucrado en la explicación de estas asociaciones es posiblemente múltiple y aún su comprensión está en fases iniciales. Una posibilidad es que la isquemia placentaria causante de un porcentaje importante de las desnutriciones uterinas induzca una vasoconstricción fetal compensatoria, la que puede programar los sistemas vasopresores fetales o incluso generar modificaciones vasculares estructurales. Otra causa de esta asociación podría ser la participación de un genotipo fetal predominantemente vasoconstrictivo, que va a generar una placentación deficiente y se expresará en la hipertensión de la vida adulta.
DIAGNÓSTICO DE DISFUNCIÓN ENDOTELIAL
La disfunción endotelial puede ser evaluada clínicamente a través de mediciones de vasodilatación dependiente de endotelio, y también por marcadores plasmáticos como endotelina 1, factor von Willebrand, trombomodulina y moléculas de adhesión de monocitos. Sin embargo ninguno de ellos por sí sólo logra reflejar la indemnidad de la función endotelial. Hasta ahora la evaluación más eficaz de la función endotelial se logra a través de la respuesta vasodilatadora a un estímulo farmacológico o mecánico. Entre los factores identificados como estimuladores de la función del endotelio se encuentran acetilcolina, sustancia P, serotonina, bradicinina y trombina, que actúan en relación a receptores de membrana adosados a sistemas de transducción de proteínas G. También el roce provocado por un aumento del flujo sanguíneo constituye un estímulo mecánico de la vasodilatación a través de liberación de óxido nítrico.
Caracterización de la disfunción endotelial. En las primeras evaluaciones de la función endotelial se estudió la dilatación de las arterias coronarias inducida por dipiridamol en sujetos portadores de angina microvascular, en los que se determinó la existencia de alteración en la respuesta vasodilatadora. La evaluación de la respuesta a la isquemia en la arteria braquial de estos sujetos también mostró una respuesta alterada, observándose correlación en el deterioro de la respuesta en ambos territorios. Los métodos para evaluar la función endotelial coronaria son invasivos y por lo tanto difíciles de repetir en forma seriada.
Actualmente se dispone de un método no invasivo utilizando ultrasonido de alta resolución para medir la dilatación mediada por flujo en arteria braquial. Anderson y cols. estudiaron 50 pacientes sometidos a cateterización en los que se comparó la respuesta vasodilatadora coronaria a acetilcolina con el cambio en el diámetro de arteria braquial en respuesta a hiperemia (a través de stress tangencial), estableciendo una correlación entre la prueba invasiva y no invasiva. Los mejores predictores de una reducción de la respuesta vasodilatadora en arteria braquial fueron la disfunción endotelial en coronarias y la presencia de cardiopatía coronaria, lo que demostró la validez del método no invasivo en pacientes en riesgo coronario.
La vasodilatación dependiente de endotelio ha sido estudiada tanto en el territorio coronario como en la circulación periférica. El cambio en el diámetro del vaso se usa como un índice de función en vasos de conductancia, mientras que cambios en el flujo se utilizan como índice en vasos de resistencia. Las mediciones clínicas más comúnmente utilizadas incluyen:
1. Coronariografía: para medir cambios cuantitativos en el diámetro de las arterias coronarias en respuesta a dosis variables de acetilcolina.
2. Pletismografía venosa del brazo: para evaluar aumento del flujo sanguíneo después de varias concentraciones de acetilcolina.
3. Ultrasonografía de arteria braquial: Se utiliza un método no invasivo de evaluación por medio de ultrasonografía de alta resolucion según método descrito. El examen se realiza en ayunas, evitando ingesta de comidas grasas en la noche anterior y fumar en las horas previas al examen. Las mediciones se realizan en una habitación con temperatura controlada a 22°C, con el sujeto en reposo, entre las 8:00 y las 10:00 AM. El brazo no dominante del sujeto permanece extendido e inmovilizado para permitir acceso a la arteria braquial con el transductor. La imagen de la arteria braquial se registra utilizando un transductor de lineal con una frecuencia de 7,5 MHz. (GAIA 8800-MT, Medison, Seul, Korea). La imagen de la arteria braquial se obtiene 3 a 7 cm por encima del pliegue antecubital, captando una vista longitudinal de la arteria en un segmento de aproximadamente 5 cm (Figura 1). La ganancia del equipo se ajusta para obtener óptima delineación de la pared arterial (interfaz). Las mediciones se coordinan al final de diástole. Se evalúa el porcentaje de cambio en el diámetro arterial con respecto al diámetro basal (promedio de dos mediciones). Luego de la determinación del diámetro basal de la arteria, se coloca un manguito de presión por debajo del nivel donde se coloca el transductor y se aplica una presión de 200 mm Hg por 5 min para luego soltar rápidamente la oclusión. Al minuto de liberada la oclusión se realizan varias mediciones del diámetro arterial observando la dilatación en respuesta a hiperemia. Luego de 15 min. de espera, se realiza una evaluación de la vasodilatación no dependiente de endotelio, inducida por nitroglicerina (0,3 µg sublingual, 3 min. post administración), para evaluar la capacidad de respuesta del músculo liso vascular a un estímulo exógeno. Las mediciones del diámetro arterial se obtienen a partir del análisis del registro digital de la imagen. Durante cada etapa del procedimiento se registra el pulso y presión arterial del sujeto.
En las Figuras 1 y 2 se muestran las imágenes basales y post estímulo de dos pacientes con disfunción endotelial en condiciones basales y bajo tratamiento.
CONCLUSIÓN
El propósito de esta revisión ha sido analizar la información actual sobre la disfunción del endotelio y mostrar la utilidad práctica de la evaluación de función endotelial por métodos no invasivos. Si bien es cierto que esta prueba de dilatación en respuesta a hiperemia está centrada en la respuesta vasodilatadora y no permite determinar otras de las funciones del endotelio que pueden encontrarse alteradas, menos aún el mecanismo subyacente, constituye una valiosa herramienta para el manejo clínico de pacientes, en los que se puede pesquizar una situación que compromete su pronóstico cardiovascular.
cabildina...

El cerebro humano, el cerebelo: tinción inmunohistoquímica para calbindina con NCL-calbindina. Nota tinción citoplasmática de las células de Purkinje y los procesos neuronales. Parafina sección.
Calbindina es una proteína de unión a calcio que pertenecen a la superfamilia de la troponina C.Funciona como un buffer de calcio citosólico y se encuentra en los islotes del cerebro, riñón, intestino y páncreas. En el cerebro normal, calbindina (28 kDa) se ha identificado en el medio de las neuronas del tamaño de la neuropilo del compartimiento de la matriz del cuerpo estriado, los acuerdos de la fibra de lana del globo pálido y las estructuras de fibra de la retícula pars de la sustancia negra. La expresión normal de calbindina se modifica en los pacientes con parálisis supranuclear progresiva, degeneración estriatal y la enfermedad de Huntington (HD). En HD, las alteraciones del pérgolas dendríticas y espinosas neuronas estriadas pueden ser visualizados mediante técnicas de inmunohistoquímica para calbindina. En los grados moderados de alta definición, cambios proliferativos se han encontrado en estas áreas y en grados severos, los cambios degenerativos se han observado. Una proporción de las células dendríticas en la zona de luz de centros germinales son también señaló como positiva para calbindina.
Calbindins
La vitamina D dependiente de calcio proteínas de unión
Información General
Dos isoformas (D28k y D29k) de la de la vitamina D dependiente de Ca-proteínas de unión llamadocalbindins se han descrito hasta ahora. Estas proteínas se expresan en las células que tienen que manejar un influjo de calcio, tales como cerebro, huesos, dientes, oído interno y otros. Calbindins se cree que regulan la actividad celular por el calcio intracellur suprimir o almacenamiento en búfer.Calbindina D28k también se conoce como CALB 1, D-28k, y CAB 27, calbindina D29k también se conoce como la calretinina, calbindina 2, CALB 2, CAL 2, y el CAB 29.
Receptores Sensibles Al Calcio
Clase de receptores de proteína G acoplados, que reacciona a distintos niveles de CALCIO extracelular. Los receptores sensibles al calcio en las glándulas paratiroides desempeñan una importante función en el mantenimiento de lahomeostasis del calcio, regulando la liberación de hormona paratiroidea. Se diferencian de las proteínas sensoras del calcio intracelular, que detectan los niveles de calcio intracelular.
Papel del receptor sensible al calcio en la fisiología de la glándula paratiroides
RESUMEN
El receptor sensible al calcio (CaSR) representa el mecanismo molecular por el cual las células paratiroideas detectar cambios en la concentración de calcio en la sangre ionizado y modular la hormona paratiroidea (PTH) para mantener los niveles séricos de calcio dentro de un estrecho rango fisiológico. Se ha aprendido mucho en los últimos años sobre la diversidad de la transducción de señales a través de la CaSR y los diversos factores que afectan la expresión del receptor.Más allá de su papel clásico como un factor determinante de calcio regulados por la secreción de PTH, la señalización a través de la CaSR también influye tanto en la transcripción de genes y la proliferación celular en las células paratiroideas. El CaSR por lo tanto sirve una amplia función fisiológica mediante la integración de varios aspectos distintos de la función de las glándulas paratiroideas. La actual revisión se resumen los acontecimientos recientes que mejoran nuestra comprensión de la CaSR y su importancia fundamental en la fisiología de la glándula paratiroides.
El calcio extracelular receptor sensible |
Mantener un estricto control sobre la concentración de calcio en la sangre y el líquido extracelular es una tarea crítica. Es lógico que un sensor de calcio se desarrollaría como un componente del sistema responsable de la homeostasis del calcio. Teniendo en cuenta su participación en los procesos fisiológicos de modulación de tantos, el calcio se puede considerar como un tipo de hormona, y el sensor de calcio como de su receptor. La secuencia de ADN que codifica para el sensor de calcio extracelular fue aislado originalmente a partir de la glándula paratiroidea bovina. Desde entonces, las secuencias correspondientes han sido aislados a partir de una amplia gama de especies, lo que permite un estudio serio de esta proteína de la membrana intrigante.  El receptor sensible al calcio es un miembro de la familia de receptores acoplados a proteínas G. Al igual que otros miembros de la familia, que contiene siete hélices hidrofóbicas que se ancla en la membrana plasmática. Los grandes (~ 600 aminoácidos) dominio extracelular se sabe que es fundamental para la interacción con el calcio extracelular. El receptor también tiene un lugar grande (~ 200 aminoácidos) cola citosólica. Estas características se muestran en la figura de la derecha, los puntos rojos en el dominio intracelular corresponden a los sitios de fosforilación de la proteína quinasa potenciales. El receptor sensible al calcio es un miembro de la familia de receptores acoplados a proteínas G. Al igual que otros miembros de la familia, que contiene siete hélices hidrofóbicas que se ancla en la membrana plasmática. Los grandes (~ 600 aminoácidos) dominio extracelular se sabe que es fundamental para la interacción con el calcio extracelular. El receptor también tiene un lugar grande (~ 200 aminoácidos) cola citosólica. Estas características se muestran en la figura de la derecha, los puntos rojos en el dominio intracelular corresponden a los sitios de fosforilación de la proteína quinasa potenciales.La activación del sensor de calcio tiene dos grandes transducción de la señal de efectos:
El sensor de calcio se expresa en una amplia gama de células, incluyendo células paratiroides y las células C de la glándula tiroides, lo que indica su participación en el control de la síntesis y secreción de la hormona paratiroidea y calcitonina . Estudios funcionales y la investigación de los animales con mutaciones en el gen sensor de calcio han confirmado que el sensor de calcio afecta directamente la secreción de estas dos hormonas. El sensor de calcio también se expresa en varios tipos de células en el riñón, los osteoblastos, una variedad de células hematopoyéticas en la médula ósea, y en la mucosa gastrointestinal. Curiosamente, también está presente en las células epiteliales escamosas del esófago. Una distribución tan amplia de expresiones admite que el concepto de que el calcio, que actúa como una hormona, tiene efectos directos sobre la función de muchos tipos de células. La comprensión del papel del sensor de calcio en la homeostasis del calcio se ha beneficiado en gran medida por el estudio de las mutaciones en el gen humano que codifica este receptor:
Final de quinta entrega.... Wilfrido Tsuchida..... |
No hay comentarios:
Los comentarios nuevos no están permitidos.