
La fenilcetonuria es un trastorno caracterizado por la defectuosa de fenilalanina en tirosina, debido a la carencia de la enzima fenilalanina hidroxilasa, por lo que la fenilalanina no puede convertida en tirosina, que es un aminoácido fundamental para el metabolismo del organismo. [1]Cuando la hidroxilasa es deficiente, se usan vías alternativas para metabolizar la fenilalanina, esta es convertida por transmutación en acido fenilpiruvio, que es reducido a acido fenilactico, o en forma, por descarboxilacion, acido fenilacteico que, por conjugación, origina la fenilcetilglutamina. Finalmente, la fenilalanina y estos productos metabólicos se acumulan en líquidos del organismo. Estos compuestos no son metabólicos anormales, sino metabólicos normales, aunque en cantidades anormales [2]Como , este aminoácido puede acumularse a niveles tóxicos en la sangre y otros tejidos, dado que las células nerviosas en el cerebro son especialmente sensibles a los niveles de fenilalanina, cantidades excesivas de esta sustancia puede causar daño cerebral, por lo que es una enfermedad progresiva severa que puede producir retraso mental si no se trata con una libre de alimentos que contengan este aminoácido a tiempo.
La incidencia de la enfermedad es de 1:10000 a 20000 personas. Se manifiesta por igual en ambos sexos. Los estudios de la genética de la población revelan claramente una herencia autonómica recesiva. La salud de los portadores no sufre efecto alguno por la presencia de este gen.[3]
Identificación y Características del gen implicado
Como se menciono anteriormente, todos los defectos se heredan con carácter autosómico recesivo. Hasta el momento se ha encontrado un gen asociado a este trastorno, conocido como el gen PAH. Este gen se encuentra en el brazo largo del cromosoma 12, entre las posiciones 22 y 24.2, abarca 90 kb y contiene 13 exones. Actualmente se han descrito más de 500 mutaciones en su cDNA, que en diferentes combinaciones, pueden dar de la gran variedad clínica y bioquímica que presentan los pacientes con deficiencia en PAH. El cromosoma 12 contiene 103.232.103 pares de bases a 103.311.380 y el gen se encuentra de los pb 103.232.103 a 103.311.380.[4]
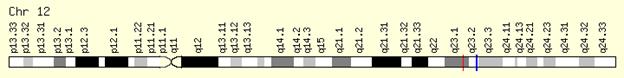
En cuanto a la identificación de este gen en 1984 Lidsky et al., demostraron que el gen PAH se encuentra en el cromosoma 12 y presumiblemente en la parte distal de 12q, por del uso de una sonda de DNAc de PAH humano para el ratón hibrido de células humanas por hibridación de Southern, la hipótesis planteada se confirmo porque en los híbridos que contienen el cromosoma translocado 12, estaban separados con PEPB (12q21) y no con TPI (12p13). [5]También en 1984 Woo et al. Asigna el gen en el cromosoma 12q21 HAP-qter mediante análisis de restricción de ADN de hámster híbridos de células somáticas humanas. Por hibridación in situ, la asignación del gen PAH se redujo a 12q22-q24.1 cromosoma[6]Por ultimo en 1985 O´Conell et al. de RFLP, confirmó la asignación del gen PAH a 12q terminal.[7]
También es importante mencionar que mediante el análisis de Nothern blot, Lichter-Konecki et al en 1999 detecto más alta expresión de un kb HAP de transcripción-2.5 en el hígado humano, seguido de riñón, páncreas y cerebro. Una transcripción de 4,6 kb se detectó también en el hígado, los riñones y el páncreas, los ensayos de RNasa confirmó HAP la expresión en el hígado y el riñón. El ARN de hibridación in situ reveló expresión de PAH en los túbulos proximales de los adultos y la corteza renal fetal y en la corteza cerebral del cerebro del feto. El análisis inmunohistoquímico confirmó expresión de la proteína PAH en túbulos renales proximales complicado.[8]
Un poco de Historia
En cuanto a la historia de la fenilcetonuria, fue en 1934 cuando Fölling realizo la descripción de una enfermedad que causaba un grave daño cerebral, en la que estaba implicada una sustancia originaria de las proteínas denominada fenilalanina; la denominó "imbecillitas phenylpyuruvica" oligofrenia fenilpirúvica, también conocida como enfermedad de Fölling[9]Tres años más tarde, Penrose y Quastel sugieren que se denomine a esta enfermedad fenilcetonuria nombre con el que se conoce hoy en . En 1947 Jervis observó que cuando se daba fenilalanina a individuos sanos, en éstos se producía un aumento de otra sustancia llamada Tirosina. Pero cuando suministraba fenilalanina a individuos fenilcetonúricos no presentaban tal elevación. Tras varios años de investigación Jervis concluyó finalmente que en la enfermedad conocida como fenilcetonuria se produce un fallo en la transformación de la fenilalanina en tirosina y que dicha transformación en los individuos sanos tiene lugar en el hígado. [10]Los avances en el tratamiento de la enfermedad no se iniciaron hasta 1953 cuando Bickel publica sus primeros trabajos en los que demuestra la efectividad de una dieta especial restringida en fenilalanina para el tratamiento de la fenilcetonuria, especulo que podría haber una relación causal entre el exceso de fenilalanina en los líquidos biológicos y una niña de daño cerebral y por lo cual planteo que sería posible mejorar su condición mediante la reducción de la ingesta de fenilalanina. El uso de un hidrolizado de caseína restringida en fenilalanina como la principal fuente de proteínas de la dieta se consideró.[11]
Mutaciones y estructura de la proteína
La proteína fenilalanina hidroxilasa tiene C-terminal del monómero PheOH que se caracteriza por el dominio catalítico, y es también responsable de tetramerización. El sitio activo PheOH se encuentra en un bolsillo profundo de las paredes helicoidal en el C-terminal. El N-terminal es el dominio de reglamentación y se extiende a través del sitio activo del dominio catalítico y es estructuralmente similar a la del dominio regulador de fosfoglicerato deshidrogenasa, una enzima implicada en la biosíntesis de serina, y PCD / DCOH, involucrados en la regeneración de BH4 y la activación de NHF1 que a su vez activa la transcripción de PAH. La siguiente figura es un monómero de PheOH, el rojo indica que el término dominio catalítico-C, el verde indica que el núcleo del ámbito regulatorio, y el azul la terminal de autorregulación de dominio-N.
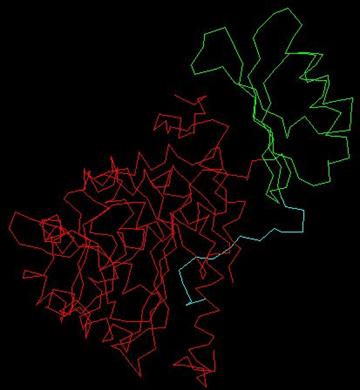
Las mutaciones en el gen PAH y sus consiguientes cambios estructurales en la enzima, dan lugar a la función PheOH reducida y la fenilcetonuria. Muchas mutaciones en el sitio activo PheOH tienen evidentes implicaciones estructurales. Sin embargo, las mutaciones en el dominio catalítico y la región N-terminal también se han identificado. Kobe et al. En anteriores postulados dicen que el N-terminal y la función reguladora de la bisagra puede ayudar a explicar la disminución de la actividad de los mutantes de PAH en las zonas que no sea el sitio activo.
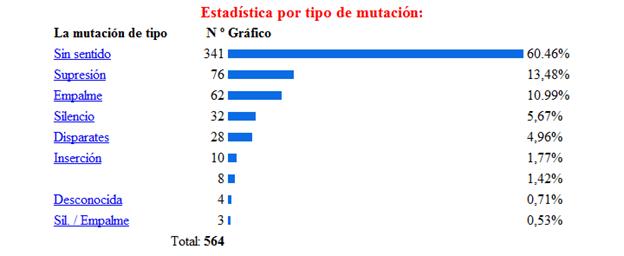
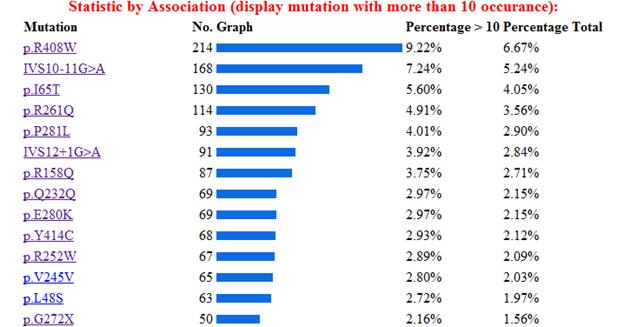
La base de la fenilcetonuria son las mutaciones producidas en el gen PAH, hasta la actualidad se han identificado 564 mutaciones en el gen PAH, según la base de datos internacional de este gen[12]Algunos estudios han establecido relaciones entre el genotipo y el fenotipo metabólico, permitiendo determinar el grado de severidad de acuerdo con la actividad de la enzima mutada, y de esta forma indirectamente también se puede afectar eldesarrollo cognitivo.[13] Los tipos de mutaciones vinculados a la PKU son la sustitución de un aminoácido por otro, deleciones, inserciones, mutaciones que alteran el procesamiento del RNA heteronuclear y las mutaciones sin sentido (etc.) En la siguiente imagen se muestra la estadística de las mutaciones más comunes del gen, estos datos fueron obtenidos de la base de datos internacional del gen PAH.[14]
De las diferentes estructuras de este gen la más afectada por las mutaciones es el exón 7 como se puede observar en la siguiente imagen donde se muestra la incidencia de las diferentes mutaciones del gen según la región apareciendo como la región más afectada el exón 7, luego el 11,10 (etc.) Lo cual se explica porque la región codificante contiene 22 sitios CPG (ricos en pares citosina-guanina), 5 de los cuales están ubicados en el exón numero siete de este gen. En el exón 7, se encuentran la mayoría de las manifestaciones que están relacionadas con hiperfenilalninemia. La hipótesis más aceptada es que esta región altamente conservada, confiere funciones esenciales para la hidroxilacion de la PAH, cualquier variación en el dominio codificado por este exón, altera la función de la enzima produciéndose la elevación de la fenilalanina. Diversas son las mutaciones que se han relaciona con la deficiencia de PAH, donde la sustitución de un aminoácido por otro es de la mas frecuéntenme encontradas.
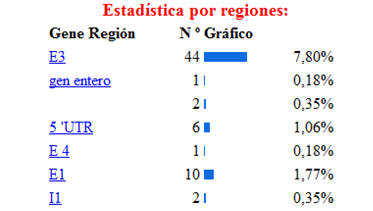
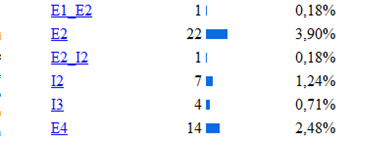
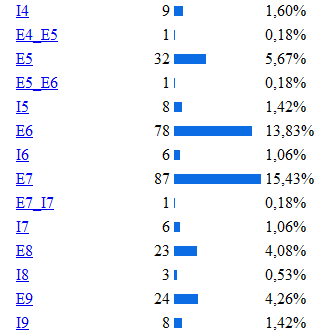
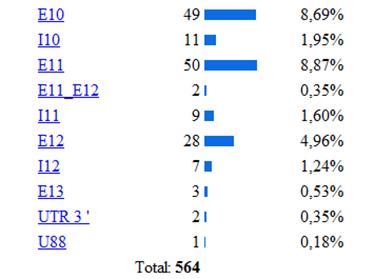
Se han clasificado 28 mutaciones como graves, como es el caso de una de las más frecuentes, la mutaciones IVS10nt-11, que en homocigosis confiere un fenotipo grave. Otro grupo de 16 mutaciones se relacionan con una actividad PAH residual con un fenotipo suave, entre las que se incluyen la 165T la V388M.
Además se han caracterizado 13 mutaciones causantes de hiperfenilalninemia benigna, como la A403V, muy frecuente en nuestra población. Se ha establecido un sistema de búsqueda sistemática de mutaciones en el gen PAH, mediante la técnica de DGGE, que permite la identificación del genotipo de los casos de PKU diagnosticados cada año en el programa de detección precoz de metabolopatias. El conocimiento del genotipo aporta al clínico unainformación esencial en la predicción de la evolución fenotípica de cada recién nacido PKU permite la aplicación de la aplicación de la dieta optima individualizada para cada paciente[15]Algunos estudios han establecido relaciones entre el genotipo y el fenotipo metabólico, permitiendo determinar el grado de severidad de acuerdo con la actividad de la enzima mutada, y de esta forma indirectamente también se puede afectar el desarrollo cognitivo.[16]
A continuación se muestra una imagen con las mutaciones más frecuentes del gen, y después una descripción de las más importantes.
IVS12DS, GA, +1
Consiste en el cambio de base individual (G/A) en el empalme de la zona donante de 5´ del intron 12[17]
En 1986 Marvit et al., encontró que el GT-a AT-sustitución en el empalme de 5´del intrón 12 dio lugar a la omisión del exón precedente, durante el empalme de ARN Se demostró que había una eliminación de 116 pares de bases internas correspondan exactamente a exón 12 y que conduce a lasíntesis de la proteína truncada que carecen de la C-terminal de 52 aminoácidos, la supresión suprimió la actividad de PAH en la célula como consecuencia de la inestabilidad de las proteínas, de hecho una sustitución de un solo nucleótido en lugar de la supresión de una base fue la base delproducto del gen anormal.[18]
GLU280LYS
La mutación consiste en un cambio de GAA/AAA en el exón 7, en la secuencia 838, esta mutación es considerada del tipo sin sentido y se encontró un cambio de glu280 a lis, identificado en un niño con una variante de fenilcetonuria. La enzima mostro actividad residual parcial, esta mutación se relacionaba con haplotipos en el sur de Europa y Norte de África. [19]
A partir del análisis de la base de datos de mutación de PAH, Byck et al en 1997 demostraron que el alelo E280K, el 1,5% de los cromosomas PKU en todo el mundo. S e presenta el 4 e haplotipos diferentes en los europeos y en haplotipos 1 y 2 en Quebec.[20]
ARG261GLN
Mutación de tipo sin sentido CGA/CAA en el exón 7, en el nucleótido 782, en el codón 261 se produce un cambio de arg a gln (R261Q). [21]Para esta mutación especifica es interesante mencionar el caso que fue observado por Zurich, Superti-Furga et al en 1992 donde observo retraso intrauterino, microcefalia, microcefalia y retraso en el desarrollo de dos primos hermanos de madres de 23 y 24 años, se encontró que había fenilalanina en concentraciones aprox de 1.2mmol/l, pero nunca había sido tratada y no presentaban retraso mental. Ambas madres demostraron ser homocigotas para mutación arg261, esto nos indica que el estado homocigoto de esta mutación se acompaña de manifestaciones clínicas leves, pero suficiente elevación de fenilalanina en la sangrepuede causar el síndrome de fenilcetonuria materna en la descendencia[22]
ARG252TRP
Hay un cambio de C-T para la transposición de la primera base en el codón 252, que da lugar a la sustitución arg252 a trp, es una mutación sin sentido descubierta por Abadie et al en 1991[23]
Se han realizado análisis de los vectores de expresión que contiene el cDNA mutantes y en los mamíferos reveló células transfectadas con insignificante actividad de la enzima, en estudios genéticos de la población italiana se mostro un marcado desequilibrio los a-trp mutación arg252 y haplotipos RFLP 1. El arg252 a trp es una mutación que se encontró en el 10% de un hapotipo de cromosomas mutantes[24]
ARG158GLN
En 1989 Dworniczak et al identifico una transición G a A (CGG/CAG) en el nucleótido 695 en el exón 5. La sustitución de base prevé un arg158 a gln (R158Q). Veinticuatro por ciento de los alelos PKU se encontraban en un contexto de haplotipo 4, todos los 7 de la a-Una transición G estaban en el haplotipo 4 de fondo. [25]
ARG243TER
Se trata de una mutación sinsentido en el exón 7 donde se produce cambio CGA/TGA en el nucleótido727 esto causa un cambio de arg243 a un codón de parada. Alelo mutante se asoció con el haplotipo 4. La mutación estaba presente en 2 de 9 haplotipo 4 alelos mutantes entre los europeos del este, pero no se encuentran entre los europeos occidentales y los asiáticos.[26]
PRO281LEU
Consiste en una mutación sin sentido donde se produce cambio de CCT/CTT, en el exón 7 en el nucleótido 842. El análisis de expresión revelo la actividad enzimática insignificante y los niveles no detectables de proteínas inmunorreactivas HAP. El pro281 a leu mutación se encontró en el 20% de un haplotipo cromosomas mutantes en la población italiana [27]
En 1994 Baric et al. Señalo que los datos que indican la mayor frecuencia de la mutación esta en Croacia, donde se detecto 55% de los alelos de un haplotipo, que corresponde al 12% de todos los alelos de PKU. Ellos interpretaron este resultado como una indicación de que la mutación se origino en el suerete de Europa[28]
TYR414CYS
Consiste en el cambio de TAC/TGC en el exón 12 en el nucleótido 1241, causando el cambio de tyr414 a CYS). La transición de Aa G en la segunda base del codón 414 fue la responsable Los estudios in vitro mostraron que la expresión-a-cis mutación tyr414 producía una proteína con una cantidad significativa de los HAP actividad de la enzima, es decir, aproximadamente el 50% del estado de equilibrio los niveles normales.[29]
En 2008 Gersting et al declaró que la mutación se produce en el Y414C produciendo dimerización del dominio de oligomerización HAP, que interactúa con el dominio catalítico de la subunidad HAP mismo. Encontraron que la tetramerización de HAP recombinante con la mutación Y414C parecía a la de la proteína de tipo salvaje. La reducción en la actividad resultante de la mutación Y414C parecía deberse a un cambio conformacional global en la proteína que reduce alosterismo.[30]
GLY272TER
Es una mutación de tipo sin sentido que causa un cambio GGA/TGA en el nucleótido 814 del exón 7 , que resulta en cambio de una gly272 a ter (G272X) de sustitución, y la deleción del codón leucina CTT 364 ( 612349.0021 ).
En 1993 Alpod et al encontró datos recopilados en la frecuencia de la mutación G272X en las poblaciones europeas. La mutación se produce al norte de los Alpes y tiene una alta frecuencia en particular en el Fiordo de La Región de Oslo de Noruega con la Bohuslän región adyacente de Suecia. Una frecuencia intermedia se observó en la parte oriental de Alemania con la parte adyacente del oeste de Checoslovaquia.Los hallazgos sugieren un origen único para esta mutación, por lo menos una población fundadora en el sureste de Noruega/Suecia adyacentes [31]
IVS10AS, GA, -11
Se produce una mutación de tipo empalme de G/A en intron 10, hay una transición en la posición 546, 11pb arriba del límite con el exón 10. La mutación se produce en un sitio de empalme crítica y da lugar a un marco de inserción, de 9 de nucleótidos entre los exones 10 y 11 del ARNm procesado. Cantidades normales de proteína en el hígado PAH estaban presentes en la PKU homocigotos de los pacientes, pero no la actividad catalítica. Esta pérdida de actividad de la enzima fue causado probablemente por cambios conformacionales que resultan de la inserción de 3 aminoácidos adicionales (Gly-Leu-Gln) entre las secuencias normales codificada por exones 10 y 11. La mutación se encuentra en estrecha asociación con los haplotipos cromosomas 6, 10 y 36. [32]Debido a la alta frecuencia de estos haplotipos en particular en Bulgaria, Italia y Turquía, Dworniczak et al. (1991) sospecha que esta mutación puede ser uno de los defectos más frecuentes en el gen que causa la PKU clásica HAP en el sur de Europa. Perez et al.(1992) también encontró esta mutación en España. Por otra parte, Pérez et al. (1993) encontraron que esta mutación es la lesión molecular predominante que causan la fenilcetonuria en Chile, Argentina y México.
Terapia Génica
El tratamiento para esta enfermedad se ha basado principalmente en el control de la dieta del paciente, pero en la actualidad se ha planteado una nueva alternativa que consiste en la terapia génica, ya había sido revelado que el método había curado fenilcetonuria (PKU) en ratones.La noticia de que esta técnica tiene efectivamente curar una enfermedad grave, aunque en ratones, ha elevado las posibilidades y las expectativas.
Además de la PKU, esta técnica podría ser usada para curar otras enfermedades genéticas causadas por falta de enzimas hepáticas como la hemofilia y las deficiencias de la enzima ciclo de la urea, así como la limpieza de colesterol de la sangre y otros.
La técnica consiste en insertar genes en regiones no codificantes del genoma lo que no hay peligro de interferencia con el funcionamiento de otros genes. Una vez insertado, el gen sigue siendo una parte permanente del genoma de la célula.
El Dr. Woo y su colega Li Chen utiliza un gen de un bacteriófago , que reconoce una secuencia específica de ADN. Esta secuencia sólo se produce varias veces en el genoma del ratón entero y siempre se encuentra en la región no codificante entre los genes.
Woo y Chen fueron capaces de curar la PKU en ratones con sólo tres inyecciones por vía intravenosa. Los niveles de fenilalanina en los ratones tratadosse redujo a valores normales y se mantuvo estable a partir de entonces.
Su color de piel también cambió de gris a negro, indicando que estaban produciendo los niveles normales de melanina, un pigmento, que está siendo producida en ratones y seres humanos con PKU.
"Debido a que los genes se insertan de forma permanente, algunas aplicaciones sería suficiente para corregir de forma permanente una enfermedad", dijo Woo.
"El reto actual es identificar un medio adecuado para la introducción de ADN en las células del hígado. Una vez que la tecnología se desarrolla, esta nueva técnica proporcionará un método seguro y eficaz de integrar el ADN en el genoma de la célula."[33]
Diagnostico, tratamiento y Pronostico
En relación a este trastorno es importante mencionar los métodos utilizados para el diagnostico, como la prueba del cloruro férrico en la orina. Existen casos raros en los que no se encuentra el ácido fenilpirúvico en la orina siempre, sólo cuando se ha expuesto al niño a una sobrecarga dietética o cuando tiene fiebre este aparece estos casos se consideran como defectos parciales.
En cuanto al tratamiento habitual comprende una dieta extremadamente baja en fenilalanina, especialmente cuando el niño está creciendo. Aquellos pacientes que continúen con la dieta hasta la vida adulta tendrán una mejor salud física y mental. La fenilalanina se encuentra en cantidades significativas en alimentos como la leche, los huevos y otros alimentos comunes. Además, se encuentra en el edulcorante artificial Nutrasweet (aspartamo), razón por la cual cualquier producto que contenga aspartamo se debe evitar.
Lofenalac es una leche en polvo infantil especial para bebés con fenilcetonuria que se puede usar durante toda la vida como fuente de proteína, con un contenido extremadamente bajo en fenilalanina y balanceada con respecto a los aminoácidos esenciales restantes.
El hecho de tomar suplementos, como el aceite de pescado, para reemplazar los ácidos grasos de cadena larga faltantes de una dieta estándar libre de fenilalanina puede ayudar a mejorar el desarrollo neurológico, incluyendo la coordinación motriz fina. Asimismo, se pueden necesitar otros suplementos específicos, como el hierro o la carnitina.
Se espera que el desenlace clínico sea muy alentador si la dieta se sigue estrictamente, comenzando poco después del nacimiento del niño; pero si el tratamiento se retrasa o el trastorno permanece sin tratamiento, se presentará daño cerebral. El desempeño escolar se puede deteriorar levemente.
Si no se evitan las proteínas que contengan fenilalanina, la fenilcetonuria puede conducir a retardo mental hacia el final del primer año de vida.
Autor:
Lina Merchancano
Universidad ICESI
16 de Mayo del 2011
No hay comentarios:
Publicar un comentario